| SOCR ≫ | DSPA ≫ | DSPA2 Topics ≫ |
DSPA2: Data Science and Predictive Analytics (UMich HS650)
Basic Visualization and Exploratory Data Analytics (Part 2)
Basic Visualization and Exploratory Data Analytics (Part 2)
SOCR/MIDAS (Ivo Dinov)
SOCR/MIDAS (Ivo Dinov)
April 2026
This is Part 2 of the larger DSPA Visualization Chapter, which is difficult to render in a single browser window due to extreme memory demands. Visualization Chapter Part 1 includes data handling, statistical measures of centrality and dispersion, understanding categorical and numeric data, uniform and normal distributions, missing data imputation, web page parsing, visualization of tabular HTML data, and cohort-rebalancing (for imbalanced groups).
In this chapter, we will present a number of complementary strategies for data wrangling, harmonization, manipulation, aggregation, visualization, and graphical exploration. Specifically, we will discuss alternative methods for loading and saving computable data objects, importing and exporting different data structures, measuring sample statistics for quantitative variables, plotting sample histograms and model distribution functions, and scraping data from websites. In addition, we will cover exploratory data analytical (EDA) techniques, handling of incomplete (missing) data, and cohort-rebalancing of imbalanced groups.
1 Exploratory Data Analytics (EDA)
In this section, we will see a broad range of simulations and hands-on activities to highlight some of the basic data visualization techniques using R. A brief discussion of alternative visualization methods is followed by demonstrations of histograms, density, pie, jitter, bar, line and scatter plots, as well as strategies for displaying trees and graphs and 3D surface plots. Many of these are also used throughout the textbook in the context of addressing the graphical needs of specific case-studies.
It is practically impossible to cover all options of every different
visualization routine. Readers are encouraged to experiment with each
visualization type, change input data and parameters, explore the
function documentation using R-help (e.g., ?plot), and
search for new R visualization packages and new functionality, which are
continuously being developed.
1.1 General Questions Driving Visualization
- What exploratory visualization techniques are available to visually interrogate my specific data?
- How to examine paired associations and correlations in a multivariate dataset?
1.2 Classification of visualization methods
Scientific data-driven or simulation-driven visualization methods are hard to classify. The following list of criteria can be used for classification:
- Data Type: structured/unstructured, small/large, complete/incomplete, time/space, ASCII/binary, Euclidean/non-Euclidean, etc.
- Task type: Task type is one of the aspects considered in classification of visualization techniques, which provides means of interaction between the researcher, the data and the display software/platform
- Scalability: Visualization techniques are subject
to some limitations, such as the amount of data that a particular
technique can exhibit
- Dimensionality: Visualization techniques can also be classified according to the number of attributes
- Positioning and Attributes: the distribution of attributes on the chart may affect the interpretation of the display representation, e.g., correlation analysis, where the relative distance among the plotted attributes is relevant for observation
- Investigative Need: the specific scientific question or exploratory interest may also determine the type of visualization:
- Examining the composition of the data
- Exploring the distribution of the data
- Contrasting or comparing several data elements, relations, association
- Unsupervised exploratory data mining.
Also, we have the following table for common data visualization methods according to task types:

We chose to introduce common data visualization methods according to this classification criterion, albeit this is not a unique or even broadly agreed upon ontological characterization of exploratory data visualization.
1.3 Composition
In this section, we will see composition plots for different types of variables and data structures.
1.3.1 Histograms and density plots
One of the first few graphs we learned in high school would be
Histogram. In R, the functions hist() or
plot_ly() represent two methods that can be applied to a
vector of values for plotting histograms. The famous 19-th century
statistician Karl
Pearson introduced histograms as graphical representations of the
distribution of a sample of numeric data. The histogram plot uses the
data to infer and display the probability distribution of the underlying
population that the data is sampled from. Histograms are constructed by
selecting a certain number of bins covering the range of values of the
observed process. Typically, the number of bins for a data array of size
\(N\) should be equal to \(\sqrt{N}\). These bins form a partition
(disjoint and covering sets) of the range. Finally, we compute the
relative frequency representing the number of observations that fall
within each bin interval. The histogram just plots a piecewise
step-function defined over the union of the bin interfaces whose height
equals the observed relative frequencies.
# Here `freq=T` shows the frequency for each *x* value and `breaks` controls for the number of bars in our histogram.
# mu <- 15; sd <- 3.7
# set.seed(1234)
# x<-rnorm(100, mean = mu, sd=sd)
# hist(x, freq=F, breaks = 10)
# lines(density(x), lwd=2, col="blue")
# t <- seq(mu-3*sd, mu+3*sd, by=0.01)
# lines(t, dnorm(t,mu,sd), col="magenta") # add the theoretical density line
library(plotly)
N <- 10000
mu <- 15; sd <- 3.7
set.seed(1234)
x <- rnorm(N, mean = mu, sd=sd)
fit <- density(x)
z<-seq(mu-4*sd, mu+4*sd, 0.1) # points from -4 to 4 in 0.1 steps
q<-seq(0.001, 0.999, 0.001) # probability quantile values from 0.1% to 99.9% in 0.1% steps
normDensity <- dnorm(z, mean=15, sd= 3.7)
plot_ly(x = x, type = "histogram", name = "Data Histogram", histnorm = "probability") %>%
add_trace(x = fit$x, y = fit$y, type = "scatter", mode = "lines", opacity=0.1,
fill = "tozeroy", yaxis = "y2", name = "Density (rnorm(100, 15, 3.7))") %>%
add_trace(x = z, y = normDensity, type = "scatter", mode = "lines", opacity=0.1,
fill = "tozeroy", yaxis = "y2", name = "Normal(15, 3.7)") %>%
layout(title='Data Histogram, Density Estimate & Theoretical Model Distribution',
yaxis2 = list(overlaying = "y", side = "right"),
legend = list(orientation = 'h'))The shape of the last histogram we draw is very close to a Normal
distribution (because we sampled from this distribution by
rnorm). Note the superposition of the corresponding Normal
density curve.
1.3.2 Pie Chart
We are all very familiar with pie charts that show us the components of a big “cake”. Although pie charts provide effective simple visualization in certain situations, it may also be difficult to compare segments within a pie chart or across different pie charts. Other plots like bar chart, box or dot plots may be attractive alternatives.
We will use the Letter Frequency Data on SOCR website to illustrate the use of pie charts.
library(rvest)
wiki_url <- read_html("https://wiki.socr.umich.edu/index.php/SOCR_LetterFrequencyData")
html_nodes(wiki_url, "#content")## {xml_nodeset (1)}
## [1] <div id="content" class="mw-body" role="main">\n\t\t\t<a id="top"></a>\n\ ...## Letter English French German
## Length:27 Min. :0.00000 Min. :0.00000 Min. :0.00000
## Class :character 1st Qu.:0.01000 1st Qu.:0.01000 1st Qu.:0.01000
## Mode :character Median :0.02000 Median :0.03000 Median :0.03000
## Mean :0.03667 Mean :0.03704 Mean :0.03741
## 3rd Qu.:0.06000 3rd Qu.:0.06500 3rd Qu.:0.05500
## Max. :0.13000 Max. :0.15000 Max. :0.17000
## Spanish Portuguese Esperanto Italian
## Min. :0.00000 Min. :0.00000 Min. :0.00000 Min. :0.00000
## 1st Qu.:0.01000 1st Qu.:0.00500 1st Qu.:0.01000 1st Qu.:0.00500
## Median :0.03000 Median :0.03000 Median :0.03000 Median :0.03000
## Mean :0.03815 Mean :0.03778 Mean :0.03704 Mean :0.03815
## 3rd Qu.:0.06000 3rd Qu.:0.05000 3rd Qu.:0.06000 3rd Qu.:0.06000
## Max. :0.14000 Max. :0.15000 Max. :0.12000 Max. :0.12000
## Turkish Swedish Polish Toki_Pona
## Min. :0.00000 Min. :0.00000 Min. :0.00000 Min. :0.00000
## 1st Qu.:0.01000 1st Qu.:0.01000 1st Qu.:0.01500 1st Qu.:0.00000
## Median :0.03000 Median :0.03000 Median :0.03000 Median :0.03000
## Mean :0.03667 Mean :0.03704 Mean :0.03704 Mean :0.03704
## 3rd Qu.:0.05500 3rd Qu.:0.05500 3rd Qu.:0.04500 3rd Qu.:0.05000
## Max. :0.12000 Max. :0.10000 Max. :0.20000 Max. :0.17000
## Dutch Avgerage
## Min. :0.00000 Min. :0.00000
## 1st Qu.:0.01000 1st Qu.:0.01000
## Median :0.02000 Median :0.03000
## Mean :0.03704 Mean :0.03741
## 3rd Qu.:0.06000 3rd Qu.:0.06000
## Max. :0.19000 Max. :0.12000We can try to plot the frequency proportion of the 26 English letters using pie and donut charts.
# The left hand side plot is the one without reference table and the right one has the table made by function `legend`.
# par(mfrow=c(1, 2))
# pie(letter$English[1:10], labels=letter$Letter[1:10], col=rainbow(10, start=0.1, end=0.8), clockwise=TRUE, main="First 10 Letters Pie Chart")
# pie(letter$English[1:10], labels=letter$Letter[1:10], col=rainbow(10, start=0.1, end=0.8), clockwise=TRUE, main="First 10 Letters Pie Chart")
# legend("topleft", legend=letter$Letter[1:10], cex=1.3, bty="n", pch=15, pt.cex=1.8, col=rainbow(10, start=0.1, end=0.8), ncol=1)
plot_ly(letter, labels = ~Letter, values = ~English, type = 'pie', name="English",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 0, column = 0)) %>%
add_pie(labels = ~Letter, values = ~Spanish, name = "Spanish",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 0, column = 1)) %>%
add_pie(labels = ~Letter, values = ~Swedish, name = "Swedish",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 1, column = 0)) %>%
add_pie(labels = ~Letter, values = ~Polish, name = "Polish",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 1, column = 1)) %>%
add_annotations(x=0.01, y=0.99,text = "English",showarrow = F, ax = 20, ay = -40) %>%
add_annotations(x=0.58, y=0.99,text = "Spanish",showarrow = F, ax = 20, ay = -40) %>%
add_annotations(x=0.01, y=0.01,text = "Swedish",showarrow = F, ax = 20, ay = -40) %>%
add_annotations(x=0.58, y=0.01,text = "Polish",showarrow = F, ax = 20, ay = -40) %>%
layout(title = 'Pie Charts of English, Spanish, Swedish & Polish Letters',
grid=list(rows=2, columns=2),
xaxis = list(showgrid = FALSE, zeroline = FALSE, showticklabels = FALSE),
yaxis = list(showgrid = FALSE, zeroline = FALSE, showticklabels = FALSE))plot_ly(letter, labels = ~Letter, values = ~German, type = 'pie', name="German",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 0, column = 0), hole = 0.5) %>%
add_pie(labels = ~Letter, values = ~Italian, name = "Italian",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 0, column = 1)) %>%
add_pie(labels = ~Letter, values = ~Dutch, name = "Dutch",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 1, column = 0)) %>%
add_pie(labels = ~Letter, values = ~Esperanto, name = "Esperanto",
textposition = 'inside', textinfo = 'label+percent', showlegend = FALSE,
domain = list(row = 1, column = 1)) %>%
add_annotations(x=0.2, y=0.78,text = "German",showarrow = F, ax = 20, ay = -40) %>%
add_annotations(x=0.8, y=0.78,text = "Italian",showarrow = F, ax = 20, ay = -40) %>%
add_annotations(x=0.2, y=0.21,text = "Dutch",showarrow = F, ax = 20, ay = -40) %>%
add_annotations(x=0.82, y=0.21,text = "Esperanto",showarrow = F, ax = 20, ay = -40) %>%
layout(title = 'Pie Charts of German, Italian, Dutch & Esperanto Letters',
grid=list(rows=2, columns=2),
xaxis = list(showgrid = FALSE, zeroline = FALSE, showticklabels = FALSE),
yaxis = list(showgrid = FALSE, zeroline = FALSE, showticklabels = FALSE))The input type for pie() is a vector of non-negative
numerical quantities. In the pie function we list the data
that we are going to use (positive and numeric), the labels for each of
them, and the colors we want to use for each sector. In the
legend function, we put the location in the first slot and
legend are the labels for colors. cex,
bty, pch, and pt.cex are all
graphic parameters that we have talked about in Chapter
1.
More elaborate pie charts, using the Latin letter data, will be
demonstrated using ggplot later, (Section
7.2.
1.3.3 Heat map
Another common data visualization method is the
heat map. Heat maps can help us visualize the individual
values in a matrix intuitively. It is widely used in genetics research
and financial applications.
We will illustrate the use of heat maps, based on a neuroimaging genetics case-study data about the association (p-values) of different brain regions of interest (ROIs) and genetic traits (SNPs) for Alzheimer’s disease (AD) patients, subjects with mild cognitive impairment (MCI), and normal controls (NC). First, let’s import the data into R. The data are 2D arrays where the rows represent different genetic SNPs, columns represent brain ROIs, and the cell values represent the strength of the SNP-ROI association as probability values (smaller p-values indicate stronger neuroimaging-genetic associations).
AD_Data <- read.table("https://umich.instructure.com/files/330387/download?download_frd=1", header=TRUE, row.names=1, sep=",", dec=".")
MCI_Data <- read.table("https://umich.instructure.com/files/330390/download?download_frd=1", header=TRUE, row.names=1, sep=",", dec=".")
NC_Data <- read.table("https://umich.instructure.com/files/330391/download?download_frd=1", header=TRUE, row.names=1, sep=",", dec=".") Then we load the R packages we need for heat maps (use
install.packages("package name") first if you did not
install them into your computer).
Then we convert the datasets into matrices.
AD_mat <- as.matrix(AD_Data); class(AD_mat) <- "numeric"
MCI_mat <- as.matrix(MCI_Data); class(MCI_mat) <- "numeric"
NC_mat <- as.matrix(NC_Data); class(NC_mat) <- "numeric"We may also want to set up the row (rc) and column (cc) colors for each cohort.
rcAD <- rainbow(nrow(AD_mat), start = 0, end = 1.0); ccAD<-rainbow(ncol(AD_mat), start = 0, end = 1.0)
rcMCI <- rainbow(nrow(MCI_mat), start = 0, end=1.0); ccMCI<-rainbow(ncol(MCI_mat), start=0, end=1.0)
rcNC <- rainbow(nrow(NC_mat), start = 0, end = 1.0); ccNC<-rainbow(ncol(NC_mat), start = 0, end = 1.0)Finally, we got to the point where we can plot heat maps. As we can
see, the input type of heatmap() is a numeric matrix.
# hvAD <- heatmap(AD_mat, col = cm.colors(256), scale = "column", RowSideColors = rcAD, ColSideColors = ccAD, margins = c(2, 2), main="AD Cohort")
# hvMCI <- heatmap(MCI_mat, col = cm.colors(256), scale = "column", RowSideColors = rcMCI, ColSideColors = ccMCI, margins = c(2, 2), main="MCI Cohort")
# hvNC <- heatmap(NC_mat, col = cm.colors(256), scale = "column", RowSideColors = rcNC, ColSideColors = ccNC, margins = c(2, 2), main="NC Cohort")
# if (!require("devtools")) install.packages("devtools")
# devtools::install_github("talgalili/d3heatmap")
# library(d3heatmap)
# d3heatmap(AD_mat, dendrogram = 'both', key = TRUE, col = 'Blues', scale = 'column', key.title = "Legend",
# print.values = T, notecol = 'white') %>%
# hmAxis("x", title = "Imaging Phenotype", location = 'bottom') %>%
# hmAxis("y", title = "Genotype", location = 'left') %>%
# hmCells(font.size = 9, color = 'blue') %>%
# hmLegend(show = T, title = "AD Cohort", location = "tl")
plot_ly(x =~colnames(AD_mat), y = ~rownames(AD_mat), z = ~AD_mat, type = "heatmap") %>%
layout(title="AD Neuroimaging-Genomic Associations (p-values)",
xaxis=list(title="ROI Imaging Biomarkers"), yaxis=list(title="SNPs"))# d3heatmap(MCI_mat, dendrogram = 'both', key = TRUE, col = 'Blues', scale = 'column', key.title = "Legend",
# print.values = T, notecol = 'white') %>%
# hmAxis("x", title = "Imaging Phenotype", location = 'bottom') %>%
# hmAxis("y", title = "Genotype", location = 'left') %>%
# hmCells(font.size = 9, color = 'blue') %>%
# hmLegend(show = T, title = "MCI Cohort", location = "tl")
plot_ly(x =~colnames(MCI_mat), y = ~rownames(MCI_mat), z = ~MCI_mat, type = "heatmap") %>%
layout(title="MCI Neuroimaging-Genomic Associations (p-values)",
xaxis=list(title="ROI Imaging Biomarkers"), yaxis=list(title="SNPs"))# d3heatmap(NC_mat, dendrogram = 'both', key = TRUE, col = 'Blues', scale = 'column', key.title = "Legend",
# print.values = T, notecol = 'white') %>%
# hmAxis("x", title = "Imaging Phenotype", location = 'bottom') %>%
# hmAxis("y", title = "Genotype", location = 'left') %>%
# hmCells(font.size = 9, color = 'blue') %>%
# hmLegend(show = T, title = "Normal Cohort", location = "tl")
plot_ly(x =~colnames(NC_mat), y = ~rownames(NC_mat), z = ~NC_mat, type = "heatmap") %>%
layout(title="(Normal) HC Neuroimaging-Genomic Associations (p-values)",
xaxis=list(title="ROI Imaging Biomarkers"), yaxis=list(title="SNPs"))In the heatmap() function the first argument is for
matrices we want to use. col is the color scheme;
scale is a character indicating if the values should be
centered and scaled in either the row direction or the column direction,
or none (“row”, “column”, and “none”); RowSideColors and
ColSideColors creates the color names for horizontal side
bars.
The differences between the AD, MCI and NC heat maps are suggestive of variations of genetic traits or alternative brain regions that may be affected in the three clinically different cohorts.
1.4 Comparison
Plots used for comparing different individuals, groups of subjects, or multiple units represent another set of popular exploratory visualization tools.
1.4.1 Paired Scatter Plots
Scatter plots use the 2D Cartesian plane to display a graph indexed
by a pair of variables. 2D points in the graph represent values
associated with the two variables corresponding to the two coordinate
axes. The position of each 2D point is determined by the values of the
first and second variables, tracked on the horizontal and vertical axes.
If no clear dependent variable exists, either variable can be plotted on
the X axis and the corresponding scatter plot will illustrate the degree
of correlation (not necessarily causation) between two variables.
Although we will mostly demonstrate the use of plot_ly(),
which provides dynamic and interactive charts, many basic graphs,
including scatter plots, can be rendered using the R function
plot(x, y).
N <- 50
ind <- c(1:N)
x<-runif(N)
y<-runif(N)
z<-runif(N)
hoverText <- paste0("Point ", ind, ": (", round(x, 3), ",", round(y, 3), ")")
# plot(x, y, main="Scatter Plot")
plot_ly(x=~x[1:20], y=~y[1:20], type="scatter", size=2, name=ind[1:20],
color=~z[1:20], mode="markers", text = hoverText[1:20]) %>%
layout(title="Random Scatterplot", xaxis=list(title="X"), yaxis=list(title="Y")) %>%
hide_colorbar()# `qplot()` is another way to plot fancy scatter plots. We can manage the colors and sizes of dots. The input type for `qplot()` is a data frame. In the following example, larger *x* will have larger dot sizes. We also grouped the data as 10 points per group.
#
# library(ggplot2)
# cat <- rep(c("A", "B", "C", "D", "E"), 10)
# plot.1 <- qplot(x, y, geom="point", size=5*x, color=cat, main="GGplot with Relative Dot Size and Color")
# print(plot.1)Now let’s draw a paired scatter plot with 5 variables.
# The input type for `pairs()` function is a matrix or data frame.
# pairs(data.frame(x, y, z))
N=1000
w<-rnorm(N)
u<-rpois(N, lambda = 1.7)
# generate some random categorical labels for all N observations
class <- sample( LETTERS[1:3], N, replace=TRUE, prob=c(0.2, 0.5, 0.3))
df <- as.data.frame(cbind(x=x,y=y,z=z,w=w,u=u, class=class))
pl_colorscale=list(c(0.0, '#19d3f3'), c(0.333, '#19d3f3'), c(0.333, '#e763fa'), c(0.666, '#e763fa'),
c(0.666, '#636efa'), c(1, '#636efa'))
axis = list(showline=FALSE, zeroline=FALSE, gridcolor='#ffff', ticklen=4)
plot_ly(df) %>%
add_trace(type = 'splom', dimensions = list( list(label='X', values=~x), list(label='Y', values=~y),
list(label='Z', values=~z), list(label='w', values=~w), list(label='U', values=~u)),
text=~class,
marker = list(color = as.integer(df$class), colorscale = pl_colorscale,
size = 7, line = list(width = 1, color = 'rgb(230,230,230)')
)
) %>%
layout(
title= 'Random Data Pairs Plot', hovermode='closest', dragmode= 'select',
plot_bgcolor='rgba(240,240,240, 0.95)',
xaxis=list(domain=NULL, showline=F, zeroline=F, gridcolor='#ffff', ticklen=4),
yaxis=list(domain=NULL, showline=F, zeroline=F, gridcolor='#ffff', ticklen=4),
xaxis2=axis, xaxis3=axis, xaxis4=axis,yaxis2=axis, yaxis3=axis, yaxis4=axis)This is an interactive scatter plot where you can select/subset some observations in any of the plots and see their associations with other variables across all pairs plots.
Let’s see a real word data example. First, we can import the Mental Health Services Survey Data into R, which is on the class website. This survey data covers \(10,374\) mental health facilities across the US, the District of Columbia, and US Territories with 237 variables about various facility characteristics. A subset of 10 variables is included in this dataset with all 10,374 cases. Two of the facilitate characteristics involve (1) supp, representing the number of specialty and support services available at the mental health facility; and (2) qual, which is the number of quality indicators present at the mental health facility.
data1 <- read.table('https://umich.instructure.com/files/399128/download?download_frd=1', header=T)
head(data1)## STFIPS majorfundtype FacilityType Ownership Focus PostTraum GLBT num
## 1 southeast 1 5 2 1 0 0 5
## 2 southeast 3 5 3 1 0 0 4
## 3 southeast 1 6 2 1 1 1 9
## 4 greatlakes NA 2 2 1 0 0 7
## 5 rockymountain 1 5 2 3 0 0 9
## 6 mideast NA 2 2 1 0 0 8
## qual supp
## 1 NA NA
## 2 15 4
## 3 15 NA
## 4 14 6
## 5 18 NA
## 6 14 NAWe can see from head() that there are a lot of
NA’s in the dataset and the pairs plot (splom)
automatically ignores these (and posts a warning message).
# plot(data1[, 9], data1[, 10], pch=20, col="red", main="qual vs supp")
# pairs(data1[, 5:10])
plot_ly(data1, x=~qual, y=~supp, type="scatter", size=2, name=STFIPS,
color=~num, mode="markers", text = STFIPS) %>%
layout(title="2010 National Mental Health Services Survey: Support Services vs. Quality Indicators Scatterplot",
xaxis=list(title="Support Services"), yaxis=list(title="Quality Indicators")) %>%
hide_colorbar()plot_ly(data1) %>%
add_trace(type = 'splom', dimensions = list( list(label='FacilityType', values=~FacilityType ),
list(label='Ownership', values=~Ownership), list(label='Focus', values=~Focus),
list(label='PostTraum', values=~PostTraum), list(label='num', values=~num)),
text=~STFIPS,
marker = list(color = as.integer(qual), colorscale = pl_colorscale,
size = 7, line = list(width = 1, color = qual)
)
) %>%
layout(
title= '2010 National Mental Health Services Survey Pairs Plot (color=qual)', hovermode='closest', dragmode= 'select',
plot_bgcolor='rgba(240,240,240, 0.95)',
xaxis=list(domain=NULL, showline=F, zeroline=F, gridcolor='#ffff', ticklen=4),
yaxis=list(domain=NULL, showline=F, zeroline=F, gridcolor='#ffff', ticklen=4),
xaxis2=axis, xaxis3=axis, xaxis4=axis,yaxis2=axis, yaxis3=axis, yaxis4=axis)The first plot shows the relation between supp (support services) and qual (quality indicators). The more elaborate pairs plot illustrates multiple bivariate relations that can be interactively explored by selecting points in any of the plots, where points are color-coded by the quality indicator variable.
To see this trend model (loess(supp ~ qual) exposing the
trajectory of the support-services to quality relationship. This
locally estimated scatterplot smoothing (LOESS) model
represents a nonlinear smoothing regression.
# plot.2 <- qplot(qual, supp, data = data1, geom = c("point", "smooth"))
# print(plot.2)
# extract only the complete cases
library(dplyr)
df1 <- data1 %>% filter_at(vars(qual,supp), all_vars(!is.na(.)))
ll.smooth = loess(df1$supp ~ df1$qual, span=0.7)
ll.pred = predict(ll.smooth, se = TRUE)
ll.df = data.frame(x=ll.smooth$x, fit=ll.pred$fit, lb=ll.pred$fit-(1.96*ll.pred$se),
ub=ll.pred$fit+(1.96*ll.pred$se))
ll.df = ll.df[order(ll.df$df1.qual),]
plot_ly(x=df1$qual, y=df1$supp, type="scatter", mode="markers", name="Data") %>%
add_lines(x=df1$qual, y=ll.pred$fit, name="Mean", line=list(color="gray", width=4)) %>%
add_ribbons(x=ll.df$df1.qual, ymin=ll.df$lb, ymax=ll.df$ub, name="95% CI",
line=list(opacity=0.4, width=1, color="lightgray")) %>%
layout(title = "LOESS Model (Supp ~ Qual) with Confidence Band",
xaxis=list(title="Quality Indicator"), yaxis=list(title="Supporting Services"))You can also use the human height and weight dataset or the knee pain dataset to illustrate some interesting scatter plots.
1.4.2 Jitter plot
Jitter plot can help us deal with the overplot issue when we have
many points in the data. The function we will be using is still in the
package ggplot2 called position_jitter().
Still we use the earthquake data for example. We will compare the
differences with and without the position_jitter()
function.
# library("xml2"); library("rvest")
wiki_url <- read_html("https://wiki.socr.umich.edu/index.php/SOCR_Data_Dinov_021708_Earthquakes")
html_nodes(wiki_url, "#content")## {xml_nodeset (1)}
## [1] <div id="content" class="mw-body" role="main">\n\t\t\t<a id="top"></a>\n\ ...earthquake <- html_table(html_nodes(wiki_url, "table")[[2]])
# plot6.1<-ggplot(earthquake, aes(Depth, Latitude, group=Magt, color=Magt))+geom_point()
# plot6.2<-ggplot(earthquake, aes(Depth, Latitude, group=Magt, color=Magt))+geom_point(position = position_jitter(w = 0.3, h = 0.3), alpha=0.5)
# print(plot6.1)
# print(plot6.2)
# Note that with option `alpha=0.5` the "crowded" places are darker than the places with only one data point.
# Sometimes, we need to add text to these points, i.e., add label in `aes` or add `geom_text`. It looks messy.
# ggplot(earthquake, aes(Depth, Latitude, group=Magt, color=Magt,label=rownames(earthquake)))+
# geom_point(position = position_jitter(w = 0.3, h = 0.3), alpha=0.5)+geom_text()
# Let's try to fix the overlap of points and labels. We need to add `check_overlap` in `geom_text` and adjust the positions of the text labels with respect to the points.
#
# ```{r warning=FALSE, message=FALSE, error=FALSE}
# ggplot(earthquake, aes(Depth, Latitude, group=Magt, color=Magt,label=rownames(earthquake)))+
# geom_point(position = position_jitter(w = 0.3, h = 0.3), alpha=0.5)+
# geom_text(check_overlap = T,vjust = 0, nudge_y = 0.5, size = 2,angle = 45)
#
# # Or you can simply use the text to denote the positions of points.
# ggplot(earthquake, aes(Depth, Latitude, group=Magt, color=Magt,label=rownames(earthquake)))+
# geom_text(check_overlap = T,vjust = 0, nudge_y = 0, size = 3,angle = 45)
# # Warning: check_overlap will not show those overlapped points. Thus, if you need an analysis at the level of every instance, do not use it.
glyphication <- function (name) {
glyph= vector()
for (i in 1:length(name)){
glyph[i]="triangle-up"
if (name[i]=="Md") { glyph[i]="diamond-open" }
else if (name[i]=="ML") { glyph[i]="circle-open" }
else if (name[i]=="Mw") { glyph[i]="square-open" }
else if (name[i]=="Mx") { glyph[i]="x-open" }
}
return(glyph)
}
earthquake$glyph <- glyphication(earthquake$Magt)
plot_ly(earthquake) %>%
add_markers(x = ~Longitude, y = ~Latitude, type = "scatter", color = ~Magt,
mode = "markers", marker = list(size = ~Depth, color = ~Magt, symbol = ~glyph,
line = list(color = ~Magt, width = 3))) %>%
layout(title="California Earthquakes (1969 - 2007)")1.4.3 Bar Plots
Bar plots, or bar charts, represent group data with rectangular bars. There are many variants of bar charts for comparison among categories. Typically, either horizontal or vertical bars are used where one of the axes shows the compared categories and the other axis represents a discrete value. It’s possible, and sometimes desirable, to plot bar graphs including bars clustered by groups.
In R we can use plotly or barplot() for
barplots with inputs either vectors or matrices. The
ggplot2::diamonds dataset is comprised of \(53,940\) diamond records (rows) with 10
observed characteristics: price ($326–$18,823); carat (diamond weight);
cut (quality of the cut); color (D (best) to J (worst)); clarity (I1
(worst), …, IF (best)); x, and z length in mm; depth (total depth
percentage = z/mean(x, y) = 2*z/(x + y)); and table (diamond width of
top).
We can add error-bars to each bar to indicate a statistical variability. T
# bar <- barplot(m <- rowMeans(x) * 10, ylim=c(0, 10))
# stdev <- sd(t(x[1:4, ]))
# arrows(bar, m, bar, m + stdev, length=0.15, angle = 90)
plot_ly(ggplot2::diamonds, y = ~log(price), color=~cut, type = "box") %>%
layout(title = "Boxplot of Diamond (log) Price by Cut",
xaxis=list(title="Diamond Cut"))plot_ly(ggplot2::diamonds, x= ~clarity, y = ~log(price), color=~color, type = "box") %>%
layout(boxmode = "group", title = "Grouped Boxplot of Diamond (log) Price by Clarity and Color",
legend=list(title=list(text='<b> Diamond Color </b>')),
xaxis=list(title="Diamond Clarity"))Let’s look at a more complex example. We utilize the dataset Case_04_ChildTrauma for illustration. This case study examines associations between post-traumatic psychopathology and service utilization by trauma-exposed children.
data2 <- read.table('https://umich.instructure.com/files/399129/download?download_frd=1', header=T)
attach(data2)
head(data2)## id sex age ses race traumatype ptsd dissoc service
## 1 1 1 6 0 black sexabuse 1 1 17
## 2 2 1 14 0 black sexabuse 0 0 12
## 3 3 0 6 0 black sexabuse 0 1 9
## 4 4 0 11 0 black sexabuse 0 1 11
## 5 5 1 7 0 black sexabuse 1 1 15
## 6 6 0 9 0 black sexabuse 1 0 6We have two character variables. Our goal is to draw a bar plot
comparing the means of age and service among
different races in this study and we want to add standard deviation for
each bar. The first thing to do is delete the two character columns.
Remember the input for barplot() is numerical vector or
matrix. However, we will need race information for classification. Thus,
we store it in a different dataset.
Then, we are ready to separate groups and get group means.
data2.df <- as.data.frame(data2)
Blacks <- data2[which(data2$race=="black"), ]
Other <- data2[which(data2$race=="other"), ]
Hispanic <- data2[which(data2$race=="hispanic"), ]
White <- data2[which(data2$race=="white"), ]
B <- c(mean(Blacks$age), mean(Blacks$service))
O <- c(mean(Other$age), mean(Other$service))
H <- c(mean(Hispanic$age), mean(Hispanic$service))
W <- c(mean(White$age), mean(White$service))
x <- cbind(B, O, H, W)
x## B O H W
## [1,] 9.165 9.12 8.67 8.950000
## [2,] 9.930 10.32 9.61 9.911667Until now, we had a numerical matrix for the means available for plotting. Now, we can compute a second order statistics - standard deviation, and plot it along with the means, to illustrate the amount of dispersion for each variable.
# bar <- barplot(x, ylim=c(0, max(x)+2.0), beside=TRUE,
# legend.text = c("age", "service") , args.legend = list(x = "right"))
# text(labels=round(as.vector(as.matrix(x)), 2), x=seq(1.4, 21, by=1.5), #y=as.vector(as.matrix(x[1:2, ]))+0.3)
# y=11.5)
#
# m <- x; stdev <- sd(t(x))
# arrows(bar, m, bar, m + stdev, length=0.15, angle = 90)
# Here, we want the y margin to be little higher than the greatest value (`ylim=c(0, max(x)+2.0)`) because we need to leave space for value labels. Now we can easily notice that Hispanic trauma-exposed children are the youngest in terms of average age and they are less likely to utilize services like primary care, emergency room, outpatient therapy, outpatient psychiatrist, etc.
# Diamonds Dataset example
# data_mean <- ddply(diamonds, c("clarity", "cut"), summarize, price = mean(price))
# data_sd <- ddply(diamonds, c("clarity", "cut"), summarize, price = sd(price))
# data2 <- data.frame(data_mean, sd=data_sd$price)
#
# plot_ly(data = data2[which(data2$cut == 'Ideal'), ], x = ~clarity, y = ~price, type = 'bar',
# name = 'Cut=Ideal', error_y = ~list(array = sd, color = '#000000')) %>%
# add_trace(data = data2[which(data2$cut == 'Premium'), ], name = 'Cut=Premium') %>%
# add_trace(data = data2[which(data2$cut == 'Very Good'), ], name = 'Cut=Very Good') %>%
# add_trace(data = data2[which(data2$cut == 'Good'), ], name = 'Cut=Good') %>%
# add_trace(data = data2[which(data2$cut == 'Fair'), ], name = 'Cut=Fair') %>%
# layout(title="Statistical Barplots (Diamonds Dataset)",
# legend=list(title=list(text='<b> Diamond Cuts </b>')))
library(plyr)
data_mean <- ddply(data2, c("traumatype", "race"), summarise, service = mean(service))
data_sd <- ddply(diamonds, c("traumatype", "race"), summarise, service = sd(service))
data2 <- data.frame(data_mean, sd=data_sd$service)
plot_ly(data = data2[which(data2$race == 'black'), ], x = ~traumatype, y = ~service, type = 'bar',
name = 'Black', error_y = ~list(array = sd, color = '#000000')) %>%
add_trace(data = data2[which(data2$race == 'hispanic'), ], name = 'Hispanic') %>%
add_trace(data = data2[which(data2$race == 'other'), ], name = 'Other') %>%
add_trace(data = data2[which(data2$race == 'white'), ], name = 'White') %>%
layout(title="Statistical Barplots (Child Trauma Dataset)",
legend=list(title=list(text='<b> Race </b>')))Another way to plot bar plots is to use ggplot() in the
ggplot package. This kind of bar plots are quite different from the one
we introduced previously. It plots the counts of character variables
rather than the means of numerical variables. It takes the values from a
data.frame. Unlike barplot(), drawing bar
plots using ggplot2 requires remaining character variables
in the original data frame.
library(ggplot2)
#data2 <- read.table('https://umich.instructure.com/files/399129/download?download_frd=1', header=T)
ggplot(data2, aes(race, fill=race)) + geom_bar()+facet_grid(. ~ traumatype) 
This plot helps us to compare the occurrence of different types of child-trauma among different races.
1.4.4 Trees and Graphs
In general, a graph is an ordered pair \(G = (V, E)\) of vertices (\(V\)). i.e., nodes or points, and a set edges (\(E\)), arcs or lines connecting pairs of nodes in \(V\). A tree is a special type of acyclic graph that does not include looping paths. Visualization of graphs is critical in many biosocial and health studies and we will see examples throughout this textbook.
In Chapter 3 and Chapter 8 we will learn more about how to build tree models and other clustering methods, and in Chapter 22, we will discuss deep learning and neural networks, which intrinsically represent AI decision graphs.
This section will be focused on displaying tree graphs. We will use 02_Nof1_Data.csv for this demonstration.
data3<- read.table("https://umich.instructure.com/files/330385/download?download_frd=1", sep=",", header = TRUE)
head(data3)## ID Day Tx SelfEff SelfEff25 WPSS SocSuppt PMss PMss3 PhyAct
## 1 1 1 1 33 8 0.97 5.00 4.03 1.03 53
## 2 1 2 1 33 8 -0.17 3.87 4.03 1.03 73
## 3 1 3 0 33 8 0.81 4.84 4.03 1.03 23
## 4 1 4 0 33 8 -0.41 3.62 4.03 1.03 36
## 5 1 5 1 33 8 0.59 4.62 4.03 1.03 21
## 6 1 6 1 33 8 -1.16 2.87 4.03 1.03 0We use hclust to build the hierarchical cluster model.
hclust takes only inputs that have dissimilarity structure
as produced by dist(). Also, we use the ave()
method for agglomeration and plot our first tree graph.

When we have no limit for maximum cluster groups, we will get the
above graph, which is miserable to look at. Luckily, cutree
will help us to set limitations to the number of clusters.
cutree() takes a hclust object and returns a
vector of group indicators for all observations.
require(graphics)
mem <- cutree(hc, k = 10)
# mem; # to print the hierarchical tree labels for each case
# which(mem==5) # to identify which cases belong to class/cluster 5
# To see the number of Subjects in which cluster:
# table(cutree(hc, k=5))Then, we can get the mean of each variable within groups by the following for loop.
Now we can plot the new tree graph with 10 groups. With
members=table(mem) option, the matrix is taken to be a
dissimilarity matrix between clusters instead of dissimilarities between
singletons and members giving the number of observations per
cluster.
hc1 <- hclust(dist(cent), method = "ave", members = table(mem))
plot(hc1, hang = -1, main = "Re-start from 10 clusters")
# via plot_ly()
# library(plotly)
# library(ggplot2)
# library(ggdendro)
# hc1$labels <- paste("Cluster", 1:10)
# p <- ggdendrogram(hc1, rotate = FALSE, size = 2)
# ggplotly(p)
library(plotly)
library(ggdendro)
# Extract data
hc1$labels <- paste("Cluster", 1:10)
dend_data <- dendro_data(hc1, type = "rectangle")
segments <- dend_data$segments
# Create plot using native plotly pipes
p <- plot_ly() %>%
add_segments(
data = segments,
x = ~x,
y = ~y,
xend = ~xend,
yend = ~yend,
line = list(color = "black"),
hoverinfo = "none"
) %>%
layout(
xaxis = list(
title = "Re-start from 10 clusters",
tickvals = seq_along(dend_data$labels$label),
ticktext = dend_data$labels$label
),
yaxis = list(title = "Height"),
showlegend = FALSE
)
p1.4.5 Correlation Plots
The corrplot package enables the graphical display of a
correlation matrix, and confidence intervals, along with some tools for
matrix reordering. There are seven visualization methods (parameter
method) in the corrplot package, named “circle”, “square”,
“ellipse”, “number”, “shade”, “color”, “pie”.
Let’s use 03_NC_SNP_ROI_Assoc_P_values.csv again to investigate the associations among SNPs using correlation plots.
The corrplot() function we will be using takes
correlation matrix only. So we need to get the correlation matrix of our
data first via the cor() function.
# install.packages("corrplot")
library(corrplot)
NC_Associations_Data <- read.table("https://umich.instructure.com/files/330391/download?download_frd=1", header=TRUE, row.names=1, sep=",", dec=".")
M <- cor(NC_Associations_Data)
M[1:10, 1:10]## P2 P5 P9 P12 P13 P14
## P2 1.00000000 -0.05976123 0.99999944 -0.05976123 0.21245299 -0.05976123
## P5 -0.05976123 1.00000000 -0.05976131 -0.02857143 0.56024640 1.00000000
## P9 0.99999944 -0.05976131 1.00000000 -0.05976131 0.21248635 -0.05976131
## P12 -0.05976123 -0.02857143 -0.05976131 1.00000000 -0.05096471 -0.02857143
## P13 0.21245299 0.56024640 0.21248635 -0.05096471 1.00000000 0.56024640
## P14 -0.05976123 1.00000000 -0.05976131 -0.02857143 0.56024640 1.00000000
## P15 -0.08574886 0.69821536 -0.08574898 -0.04099594 0.36613665 0.69821536
## P16 -0.08574886 0.69821536 -0.08574898 -0.04099594 0.36613665 0.69821536
## P17 -0.05976123 -0.02857143 -0.05976131 -0.02857143 -0.05096471 -0.02857143
## P18 -0.05976123 -0.02857143 -0.05976131 -0.02857143 -0.05096471 -0.02857143
## P15 P16 P17 P18
## P2 -0.08574886 -0.08574886 -0.05976123 -0.05976123
## P5 0.69821536 0.69821536 -0.02857143 -0.02857143
## P9 -0.08574898 -0.08574898 -0.05976131 -0.05976131
## P12 -0.04099594 -0.04099594 -0.02857143 -0.02857143
## P13 0.36613665 0.36613665 -0.05096471 -0.05096471
## P14 0.69821536 0.69821536 -0.02857143 -0.02857143
## P15 1.00000000 1.00000000 -0.04099594 -0.04099594
## P16 1.00000000 1.00000000 -0.04099594 -0.04099594
## P17 -0.04099594 -0.04099594 1.00000000 -0.02857143
## P18 -0.04099594 -0.04099594 -0.02857143 1.00000000We will discover the difference among different methods under
corrplot.

# par specs c(bottom, left, top, right) which gives the margin size specified in inches
corrplot(M, method = "square", title = "square", tl.cex = 0.5, tl.col = 'black', mar=c(1, 1, 1, 1))
corrplot(M, method = "ellipse", title = "ellipse", tl.cex = 0.5, tl.col = 'black', mar=c(1, 1, 1, 1))

corrplot(M, type = "upper", tl.pos = "td",
method = "circle", tl.cex = 0.5, tl.col = 'black',
order = "hclust", diag = FALSE, mar=c(1, 1, 0, 1))

The shades are different and darker dots represent high correlation of the two variables corresponding to the x and y axes.
1.5 Relationships
1.5.1 Line plots using
ggplot
Line charts display a series of data points, e.g., observed intensities (\(Y\)) over time (\(X\)), by connecting them with straight-line segments. These can be used to either track temporal changes of a process or compare the trajectories of multiple cases, time series or subjects over time, space, or state.
In this section, we will utilize the Earthquakes dataset on SOCR website. It records information about earthquakes that occurred between 1969 and 2007 with magnitudes larger than 5 on the Richter scale.
# library("xml2"); library("rvest")
wiki_url <- read_html("https://wiki.socr.umich.edu/index.php/SOCR_Data_Dinov_021708_Earthquakes")
html_nodes(wiki_url, "#content")## {xml_nodeset (1)}
## [1] <div id="content" class="mw-body" role="main">\n\t\t\t<a id="top"></a>\n\ ...In this dataset, we set Magt(magnitude type) as groups.
We will draw a “Depth vs Latitude” line plot from this dataset. The
function we are using is called ggplot() under
ggplot2. The input type for this function is mostly data
frame and aes() specifies aesthetic mappings of how
variables in the data are mapped to visual properties (aesthetics) of
the geom objects, e.g., lines.
library(ggplot2)
plot4 <- ggplot(earthquake, aes(Longitude, Latitude, group=Magt, color=Magt))+
# Either draw lines
# geom_line()
# or, alternatively, we can draw glyphs/points
geom_point(data=earthquake, size=4, mapping=aes(x=Longitude, y=Latitude, shape=Magt))
plot4 # or print(plot4)
The first part
ggplot(earthquake, aes(Depth, Latitude, group=Magt, color=Magt))
in the code specifies the setting of the plot: dataset, group and color.
The second part specifies we are going to draw (points or) lines between
data points. In later chapters, we will frequently use the package
ggplot2 and the structure under this great package is
always function1+function2.
1.5.2 Density Plots
We can visualize the distribution for different variables using density plots.
The following segment of R code plots the distribution for latitude
among different earthquake
magnitude types. Also, it is using the ggplot()
function but combined with geom_density().
# library("ggplot2")
ggplot(earthquake, aes(Latitude, group=Magt, newsize=2))+geom_density(aes(color=Magt), size = 2) +
theme(legend.position = 'right',
legend.text = element_text(color= 'black', size = 12, face = 'bold'),
legend.key = element_rect(size = 0.5, linetype='solid'),
legend.key.size = unit(1.5, 'lines'))
Note how the green magt type (Local (ML) earthquakes)
has a peak at latitude \(37.5\), which
represents 37-38 degrees
North.
1.6 Distributions
Recall that there is a duality between theoretical and empirical mass, density, and distribution functions. Earlier, we saw the relations between these using the (continuous) Normal distribution, let’s now look at the (discrete) Poisson distribution. The graph below plots (1) the histogram of a sample of 1,000 Poisson(1) random observations (light blue color), (2) the theoretical density/mass function (magenta color), and (3) a smooth continuous (Gaussian) kernel density estimation based on the random sample (blue color). More interactive plots of univariate distributions and multivariate distributions are available online.
set.seed(1234)
poisson_sample <- rpois(1000, 1)
# slightly offset the histogram bins to align with mass function
hist_breakes <- c(-0.5, 0.5, 1.5, 2.5, 3.5, 6.5)
# hist(poisson_sample, freq=F, breaks = hist_breakes, col="light blue", lwd=2, ylim = c(0, 0.45))
# lines(density(poisson_sample, kernel = "gaussian"), lwd=2, col="blue")
# t <- seq(0, 6, by=0.01)
# lines(t, dpois(t,1), type="h", col="magenta", lwd=6) # add the theoretical density line
# legend(3,0.3, legend=c("Sample histogram (n=1,000)", "Theoretical mass function",
# "Gaussian kernel density estimate"),
# bty = "n", box.lty=0, col=c("light blue", "magenta", "blue"), lty=1, lwd=3)
h <-hist(poisson_sample, breaks = hist_breakes, plot = F)
t <- seq(0, 6, by=0.01)
Pois <- density(poisson_sample, kernel = "gaussian")
plot_ly(x = h$mids, y = h$density, type = "bar", name="Sample Histogram") %>%
add_lines(x=t, y=dpois(t,1), type="scatter", mode="lines",
name="(Theoretical) Poisson Mass Function") %>%
add_lines(x=Pois$x, y=Pois$y,
type="scatter", mode="lines",
name="Gaussian kernel density estimate (sample)") %>%
layout(bargap=0.1, title="Histogram (Simulated Poisson Data)",
legend = list(orientation = 'h'))1.6.1 Data Modeler
A common task in data-driven inference involves the fitting of appropriate distribution models to specific observed data elements (features). In general, as there are uncountably many possible distributions that can be used as models for various types of processes, this is a difficult task. The Probability Distributome Project (see Distributome Navigator) provides a deeper understanding of the notion of a probability distribution and the relations between various distributions.
We will demonstrate the concept of a data modeler by
using crystallographic
data from the Ivanova Lab
at the University of Michigan, which includes the crystal spectra of
9
length samples and 9
width samples. For both, the length and width spectra, the 9
features include “AC1338”, “AC1432”, “AC1593”, “AC1679”, “AC1860”,
“AC1874”, “AC1881”, “AC1903”, and “Rec” (these represent different
samples). Notice that the nine spectra are not congruent, different
features have different sampling rates. We will employ the fitdistrplus
R-package to estimate the parameters of 3 complementary
distributions, however, there are many alternative packages that can
also be used.
1.6.1.1 Loading the spectral crystallography data
The data include two separate signals capturing the spectral length and the width of the crystallographic sample.
- Dec 2019 crystallography spectral data
- crystallography Length data are here
- crystallography Width data are here
# You may choose which of the 2 CSV files (width or length) to work with
crystallography_Length_data <- read.csv(file = "https://umich.instructure.com/files/11653615/download?download_frd=1",
header=TRUE)
crystallography_Width_data <- read.csv(file = "https://umich.instructure.com/files/11653614/download?download_frd=1",
header=TRUE)
crystallography_data <- crystallography_Length_data
# crystallography_data <- crystallography_Width_data
# Get the feature names (IDs)
colNames <- colnames(crystallography_data); colNames## [1] "AC1338" "AC1432" "AC1593" "AC1679" "AC1860" "AC1874" "AC1881" "AC1903"
## [9] "Rec"1.6.1.2 Feature distributions
Let’s plot the histograms of each of the nine features.
# plot all histograms
library(tidyr)
# library(ggplot2)
# # or `library(tidyverse)`
#
# crystallography_data %>% gather() %>% head()
# # key value
# #1 AC1338 70.547
# #2 AC1338 40.448
# #3 AC1338 47.212
# #4 AC1338 91.468
# #5 AC1338 79.088
# #6 AC1338 132.319
# #...
# crystallography_data %>% gather() %>% tail()
# # key value
# #5872 Rec 68.479
# #5873 Rec 41.047
# #5874 Rec 47.546
# #5875 Rec 98.558
# #5876 Rec 52.956
# #5877 Rec 82.470
#
# ggplot(gather(crystallography_data), aes(value)) +
# geom_histogram(bins = 20) +
# facet_wrap(~key, scales = 'free_x')
crystalCompleteData <- crystallography_data[complete.cases(crystallography_data), ]
df_crystal <- apply(crystalCompleteData, 2, density, kernel="gaussian", bw=15)
df <- data.frame(x = unlist(lapply(df_crystal, "[[", "x")),
y = unlist(lapply(df_crystal, "[[", "y")),
sample = rep(names(df_crystal), each = length(df_crystal[[1]]$x)))
plot_ly(df, x = ~x, y = ~y, color = ~sample, type = "scatter", mode = "lines") %>%
layout(title='Crystallography Sample Densities',
legend=list(title=list(text='<b> Samples </b>')),
xaxis=list(title='X'), yaxis=list(title='Density'))1.6.1.3 Fitting single-feature univariate distribution models
We will fit Weibull, Gamma, and Log-Normal distribution models to each feature in the data.
# install.packages("fitdistrplus")
library(fitdistrplus)
col_num <- dim(crystallography_data)[2]; col_num## [1] 9# Store the Weibull, Gamma, and Log-Normal Distribution models for the 9 features
fit_W <- vector(mode = "list", length = col_num)
fit_G <- vector(mode = "list", length = col_num)
fit_LN <- vector(mode = "list", length = col_num)
for(i in 1:col_num) {
data_no_NA <- crystallography_data[complete.cases(crystallography_data[, i]), i]
length(data_no_NA)
fit_W[[i]] <- fitdist(data_no_NA, "weibull"); summary(fit_W[i])
fit_G[[i]] <- fitdist(data_no_NA, "gamma"); summary(fit_G[i])
fit_LN[[i]] <- fitdist(data_no_NA, "lnorm"); summary(fit_LN[i])
}
# extract the model parameters
W_mod_p1_name = array(dim=c(col_num,2)); dim(W_mod_p1_name) # param name## [1] 9 2## [1] 9 2## [1] 9 2## [1] 9 2## [1] 9 2## [1] 9 2# Compute the mean (m) and standard deviation (sd) for each model distribution
W_mod_mean = array(dim=c(col_num,1)); length(W_mod_mean) # Weibull mean or mode## [1] 9## [1] 9## [1] 9## [1] 9## [1] 9## [1] 9for(i in 1:col_num) {
W_mod_p1_name[i, 1] <- names(fit_W[[i]]$estimate[1]) # Weibull "shape"
W_mod_p1_val[i, 1] <- fit_W[[i]]$estimate[[1]]
W_mod_p1_name[i, 2] <- names(fit_W[[i]]$estimate[2]) # Weibull "scale"
W_mod_p1_val[i, 2] <- fit_W[[i]]$estimate[[2]]
W_mod_mean[i] = W_mod_p1_val[i, 2] * gamma(1+1/W_mod_p1_val[i, 1]) # Weibull mean
W_mod_mean[i] = W_mod_p1_val[i, 2] *
((W_mod_p1_val[i, 1]-1)/W_mod_p1_val[i, 1])^(1/W_mod_p1_val[i, 1]) # Weibull mode
W_mod_sd[i] = W_mod_p1_val[i, 2]*sqrt(gamma(1+2/W_mod_p1_val[i, 1])-
(gamma(1+1/W_mod_p1_val[i, 1]))^2) # Weibull SD
G_mod_p1_name[i, 1] <- names(fit_G[[i]]$estimate[1]) # Gamma "shape"
G_mod_p1_val[i, 1] <- fit_G[[i]]$estimate[[1]]
G_mod_p1_name[i, 2] <- names(fit_G[[i]]$estimate[2]) # Gamma "scale"
G_mod_p1_val[i, 2] <- fit_G[[i]]$estimate[[2]]
G_mod_mean[i] = G_mod_p1_val[i, 1] / G_mod_p1_val[i, 2] # Gamma mean
G_mod_mean[i] = (G_mod_p1_val[i, 1]-1) / G_mod_p1_val[i, 2] # Gamma mode
G_mod_sd[i] = sqrt(G_mod_p1_val[i, 1]) / G_mod_p1_val[i, 2] # Gamma SD
LN_mod_p1_name[i, 1] <- names(fit_LN[[i]]$estimate[1]) # Log-normal "shape"
LN_mod_p1_val[i, 1] <- fit_LN[[i]]$estimate[[1]]
LN_mod_p1_name[i, 2] <- names(fit_LN[[i]]$estimate[2]) # Log-normal "scale"
LN_mod_p1_val[i, 2] <- fit_LN[[i]]$estimate[[2]]
LN_mod_mean[i] = exp(LN_mod_p1_val[i, 1]+ (LN_mod_p1_val[i, 2])^2/2) # Log-normal mean
LN_mod_mean[i] = exp(LN_mod_p1_val[i, 1] - LN_mod_p1_val[i, 2]^2) # Log-normal mean
LN_mod_sd[i] = sqrt((exp(LN_mod_p1_val[i, 2]^2)-1)*
exp(2*LN_mod_p1_val[i, 1]+LN_mod_p1_val[i, 2]^2)) # Log-normal SD
}
# Check results, just for one model
str(fit_W[[1]])## List of 17
## $ estimate : Named num [1:2] 2.12 96.21
## ..- attr(*, "names")= chr [1:2] "shape" "scale"
## $ method : chr "mle"
## $ sd : Named num [1:2] 0.074 2.251
## ..- attr(*, "names")= chr [1:2] "shape" "scale"
## $ cor : num [1:2, 1:2] 1 0.328 0.328 1
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:2] "shape" "scale"
## .. ..$ : chr [1:2] "shape" "scale"
## $ vcov : num [1:2, 1:2] 0.00548 0.05464 0.05464 5.06895
## ..- attr(*, "dimnames")=List of 2
## .. ..$ : chr [1:2] "shape" "scale"
## .. ..$ : chr [1:2] "shape" "scale"
## $ loglik : num -2308
## $ aic : num 4621
## $ bic : num 4629
## $ n : int 453
## $ data : num [1:453] 70.5 40.4 47.2 91.5 79.1 ...
## $ distname : chr "weibull"
## $ fix.arg : NULL
## $ fix.arg.fun: NULL
## $ dots : NULL
## $ convergence: int 0
## $ discrete : logi FALSE
## $ weights : NULL
## - attr(*, "class")= chr "fitdist"1.6.1.4 Visual inspection
Let’s examine graphically the quality of the fitted distribution models. We’ll plot the histograms of the features, the fitted probability densities, and the corresponding cumulative distribution functions (CDF) and compare them to their sample counterparts.
windows(width=20, height=8)
par(mfrow=c(3,3))
for(i in 1:col_num) {
# W_mod_p1_name[i] <- names(fit_W[[i]]$estimate[1])
# W_mod_p1_val[i] <- fit_W[[1]]$estimate[[1]]
plot.legend <- c(sprintf("Weibull(%s=%s,%s=%s) (m=%s,sd=%s)",
W_mod_p1_name[i, 1], format(W_mod_p1_val[i, 1], digits=2),
W_mod_p1_name[i, 2], format(W_mod_p1_val[i, 2], digits=2),
format(W_mod_mean[i], digits=2),
format(W_mod_sd[i], digits=2)),
sprintf("Gamma(%s=%s,%s=%s) (m=%s,sd=%s)",
G_mod_p1_name[i, 1], format(G_mod_p1_val[i, 1], digits=2),
G_mod_p1_name[i, 2], format(G_mod_p1_val[i, 2], digits=2),
format(G_mod_mean[i], digits=2),
format(G_mod_sd[i], digits=2)),
sprintf("Log-normal(%s=%s,%s=%s) (m=%s,sd=%s)",
LN_mod_p1_name[i, 1], format(LN_mod_p1_val[i, 1], digits=2),
LN_mod_p1_name[i, 2], format(LN_mod_p1_val[i, 2], digits=2),
format(LN_mod_mean[i], digits=2),
format(LN_mod_sd[i], digits=2)))
denscomp(list(fit_W[[i]], fit_G[[i]], fit_LN[[i]]), legendtext = plot.legend,
xlegend = "topright", ylegend ="right",
main=sprintf("Width: Feature: %s: Histogram & Model Densities", colnames(crystallography_data)[i]))
abline(v = format(W_mod_mean[i], digits=2), col = "red", lty=1)
abline(v = format(G_mod_mean[i], digits=2), col = "green", lty=2)
abline(v = format(LN_mod_mean[i], digits=2), col = "blue", lty=3)
# cdfcomp (list(fit_w, fit_g, fit_ln), legendtext = plot.legend)
# qqcomp (list(fit_w, fit_g, fit_ln), legendtext = plot.legend)
# ppcomp (list(fit_w, fit_g, fit_ln), legendtext = plot.legend)
}
# Plot histograms and CDF (cumulative distribution function) models
windows(width=20, height=12)
par(mfrow=c(3,3))
for(i in 1:col_num) {
plot.legend <- c(sprintf("Weibull(%s=%s,%s=%s) (m=%s,sd=%s)",
W_mod_p1_name[i, 1], format(W_mod_p1_val[i, 1], digits=2),
W_mod_p1_name[i, 2], format(W_mod_p1_val[i, 2], digits=2),
format(W_mod_mean[i], digits=2),
format(W_mod_sd[i], digits=2)),
sprintf("Gamma(%s=%s,%s=%s) (m=%s,sd=%s)",
G_mod_p1_name[i, 1], format(G_mod_p1_val[i, 1], digits=2),
G_mod_p1_name[i, 2], format(G_mod_p1_val[i, 2], digits=2),
format(G_mod_mean[i], digits=2),
format(G_mod_sd[i], digits=2)),
sprintf("Log-normal(%s=%s,%s=%s) (m=%s,sd=%s)",
LN_mod_p1_name[i, 1], format(LN_mod_p1_val[i, 1], digits=2),
LN_mod_p1_name[i, 2], format(LN_mod_p1_val[i, 2], digits=2),
format(LN_mod_mean[i], digits=2),
format(LN_mod_sd[i], digits=2)))
cdfcomp(list(fit_W[[i]], fit_G[[i]], fit_LN[[i]]), legendtext = plot.legend,
xlegend = "bottomright", ylegend ="right",
main=sprintf("Width: Feature: %s: Aggregate Hist & Model CDFs", colnames(crystallography_data)[i]))
}Below is the plot_ly() version of the model fit for one
case.
pl_list <- list()
for(i in 1:col_num) {
# W_mod_p1_name[i] <- names(fit_W[[i]]$estimate[1])
# W_mod_p1_val[i] <- fit_W[[1]]$estimate[[1]]
plot.legend <- c(sprintf("Weibull(%s=%s,%s=%s) (m=%s,sd=%s)",
W_mod_p1_name[i, 1], format(W_mod_p1_val[i, 1], digits=2),
W_mod_p1_name[i, 2], format(W_mod_p1_val[i, 2], digits=2),
format(W_mod_mean[i], digits=2),
format(W_mod_sd[i], digits=2)),
sprintf("Gamma(%s=%s,%s=%s) (m=%s,sd=%s)",
G_mod_p1_name[i, 1], format(G_mod_p1_val[i, 1], digits=2),
G_mod_p1_name[i, 2], format(G_mod_p1_val[i, 2], digits=2),
format(G_mod_mean[i], digits=2),
format(G_mod_sd[i], digits=2)),
sprintf("Log-normal(%s=%s,%s=%s) (m=%s,sd=%s)",
LN_mod_p1_name[i, 1], format(LN_mod_p1_val[i, 1], digits=2),
LN_mod_p1_name[i, 2], format(LN_mod_p1_val[i, 2], digits=2),
format(LN_mod_mean[i], digits=2),
format(LN_mod_sd[i], digits=2)))
# x <- dweibull(10000, shape=fit_W[[i]]$estimate[1], scale =fit_W[[i]]$estimate[2])
# fit <- density(x)
z <- seq(from=min(fit_W[[i]]$data), max(fit_W[[i]]$data), 0.1) # points from -4 to 4 in 0.1 steps
weibullDens <- dweibull(z, shape=fit_W[[i]]$estimate[1], scale =fit_W[[i]]$estimate[2])
gammaDens <- dgamma(z, shape=fit_G[[i]]$estimate[1], rate =fit_G[[i]]$estimate[2])
logNormalDens <- dlnorm(z, meanlog=fit_LN[[i]]$estimate[1], sdlog =fit_LN[[i]]$estimate[2])
# z<-seq(from=min(fit_W[[i]]$data), to=max(fit_W[[i]]$data), 0.1) # Range points in 0.1 steps
pl_list[[i]] <-
plot_ly(x=~fit_W[[i]]$data, name=~colnames(crystallography_data)[i], showlegend = FALSE,
marker = list(color = "transparent", line = list(color = "darkgray", width = 2)),
type="histogram", mode="markers", opacity=0.9, nbinsx=20, histnorm="probability") %>%
# add models
add_trace(x=z, y=15*weibullDens, type="scatter", mode="lines", opacity=0.5, name=plot.legend[1],
line = list(color = "red", width = 2)) %>%
add_trace(x=z, y=15*gammaDens, type="scatter", mode="lines", opacity=0.5, name=plot.legend[2],
line = list(color = "green", width = 2)) %>%
add_trace(x=z, y=15*logNormalDens, type="scatter", mode="lines", opacity=0.5, name=plot.legend[3],
line = list(color = "blue", width = 2)) %>%
# add vertical mean lines
add_segments(x=W_mod_mean[i], y=0, xend=W_mod_mean[i], yend=0.2, name="Weibull mean", color="red") %>%
add_segments(x=G_mod_mean[i], y=0, xend=G_mod_mean[i], yend=0.2, name="Gamma mean", color="green") %>%
add_segments(x=LN_mod_mean[i], y=0, xend=LN_mod_mean[i], yend=0.2, name="LogNormal mean", color="blue") %>%
layout(title = sprintf("Width: Feature: %s: Histogram & Model Densities", colnames(crystallography_data)[i]),
xaxis = list(title = colnames(crystallography_data)[i]), yaxis = list(title = "Density"),
bargap=0.1) %>% hide_colorbar()
}
pl_list %>% plotly::subplot(nrows = 3) %>% layout(title="Mixture Modeling of Crystallography Data (Interactive Plot)") 1.6.1.5 Quantitative summaries
Often, it’s useful to export the numerical results of the models. This may include various distribution characteristics like measure of centrality (e.g., mean, median, mode), measures of dispersion, and metrics of the model performance (e.g., Kolmogorov-Smirnov test).
# Save the summary outputs (mode & SD) across 9 samples, 3 models and 2 measures into a dataframe
df_matrix = array(dim=c(col_num,3*2*2)); dim(df_matrix) ## [1] 9 12for(i in 1:col_num) {
data1 <- crystallography_data[complete.cases(crystallography_data[, i]), i]
df_matrix[i, 1] = format(W_mod_mean[i], digits=2) # Weibull mode
df_matrix[i, 2] = format(W_mod_sd[i], digits=2) # Weibull SD
ks_W <- ks.test(data1, "pweibull", scale=W_mod_p1_val[i, 2], shape=W_mod_p1_val[i, 1])
df_matrix[i, 3] = format(ks_W$statistic[[1]], digits=4) # KS-test-stat Weibull
df_matrix[i, 4] = format(ks_W$p.value, digits=5) # KS-test-p-value Weibull
df_matrix[i, 5] = format(G_mod_mean[i], digits=2) # Gamma mode
df_matrix[i, 6] = format(G_mod_sd[i], digits=2) # Gamma SD
ks_G <- ks.test(data1, "pgamma", rate=G_mod_p1_val[i, 2], shape=G_mod_p1_val[i, 1])
df_matrix[i, 7] = format(ks_G$statistic[[1]], digits=4) # KS-test-stat Gamma
df_matrix[i, 8] = format(ks_G$p.value, digits=5) # KS-test-p-value Gamma
df_matrix[i, 9] = format(LN_mod_mean[i], digits=2) # Log-normal mode
df_matrix[i, 10] = format(LN_mod_sd[i], digits=2) # Log-normal SD
ks_LN <- ks.test(data1, "plnorm", sdlog=LN_mod_p1_val[i, 2], meanlog=LN_mod_p1_val[i, 1])
df_matrix[i, 11] = format(ks_LN$statistic[[1]], digits=4) # KS-test-stat Log-normal
df_matrix[i, 12] = format(ks_G$p.value, digits=5) # KS-test-p-value Log-normal
}
df_summary <- as.data.frame(df_matrix, row.names=colNames)
colnames(df_summary) <- c("Weibull_mode", "Weibull_sd","Weibull_KS.test.stat", "Weibull_KS.p.val",
"Gamma_mode", "Gamma_sd","Gamma_KS.test.stat", "Gamma_KS.p.val",
"Lognormal_mode", "Lognormal_sd","Lognormal_KS.test.stat", "Lognormal_KS.p.val")
df_summary## Weibull_mode Weibull_sd Weibull_KS.test.stat Weibull_KS.p.val Gamma_mode
## AC1338 71 42 0.0411 0.4284 64
## AC1432 75 40 0.07218 0.047982 69
## AC1593 81 54 0.05572 0.10341 75
## AC1679 81 49 0.0462 0.36208 73
## AC1860 78 45 0.06798 0.088752 73
## AC1874 75 42 0.06495 0.032324 68
## AC1881 72 58 0.0821 0.00069318 70
## AC1903 80 48 0.07426 0.059275 73
## Rec 76 41 0.05729 0.027524 68
## Gamma_sd Gamma_KS.test.stat Gamma_KS.p.val Lognormal_mode Lognormal_sd
## AC1338 42 0.02878 0.84738 57 48
## AC1432 38 0.03942 0.63424 63 40
## AC1593 52 0.03823 0.4885 67 58
## AC1679 49 0.03222 0.80172 64 56
## AC1860 42 0.03691 0.74826 67 45
## AC1874 41 0.03431 0.61239 61 45
## AC1881 55 0.05289 0.073267 63 60
## AC1903 47 0.06417 0.14456 66 51
## Rec 40 0.03865 0.28357 62 44
## Lognormal_KS.test.stat Lognormal_KS.p.val
## AC1338 0.05412 0.84738
## AC1432 0.0315 0.63424
## AC1593 0.03584 0.4885
## AC1679 0.03622 0.80172
## AC1860 0.03832 0.74826
## AC1874 0.03334 0.61239
## AC1881 0.0294 0.073267
## AC1903 0.04493 0.14456
## Rec 0.03565 0.283571.6.1.6 Mixture distribution data modeling
Earlier, we discussed the expectations maximization (EM) algorithm for parameter estimation. Now, we will illustrate the use of EM to estimate the mixture weights and the distribution parameters needed to obtain mixture-distribution data models.
For each sample, we fit a mixture distribution of \(k=3\) (different number of distribution models, which is predefined). The specific types of mixtures for each of the 9 samples are indicated below.
1.6.1.7 Mixture-distribution model fitting and parameter estimation
We will use the R package mixtools to obtain the EM estimates of the mixture distribution weights and the corresponding distribution parameters.
# crystallography_data <- read.csv(file = "https://umich.instructure.com/files/13375767/download?download_frd=1",
# header=TRUE)
# crystallography_data <- read.csv(file = "https://umich.instructure.com/files/11653615/download?download_frd=1",
# header=TRUE)
# install.packages("mixtools")
library(mixtools)
col_num <- dim(crystallography_data)[2]; col_num## [1] 9# Fit mixture models
capture.output(
for(i in 1:col_num) { # remove all non-numeric elements (if any)
# data_no_NA <- unlist(Filter(is.numeric, crystallography_data[complete.cases(crystallography_data[, i]), i]))
data_no_NA <- crystallography_data[complete.cases(crystallography_data[, i]), i]
length(data_no_NA)
fit_W[[i]] <- weibullRMM_SEM(data_no_NA, k=df_sampleMixtureParam[1,i], verb=F)
# summary(fit_W[i])
fit_G[[i]] <- gammamixEM(data_no_NA, k=df_sampleMixtureParam[1,i], verb=F)
# summary(fit_G[i])
fit_LN[[i]] <- normalmixEM(data_no_NA, k=df_sampleMixtureParam[1,i], verb=F)
# summary(fit_LN[i])
},
file='NUL'
)
# plot(fit_LN[[1]], which=2)
# lines(density(crystallography_data[complete.cases(crystallography_data[, 1]), 1]), lty=2, lwd=2)1.6.1.8 Plotting the mixture distribution models
We will define custom plots for the mixtures of Gamma,
Weibull, and Normal distributions. Alternatively, we
can also use some of the mixtools::plot() function to
display mixture distribution models.
# Custom design of Gamma-Mixture Model plot
gammaMM.plot <- function(mix.object, k = 2, main = "") { # mix.object <- fit_G[[i]]
data_no_NA <- crystallography_data[complete.cases(crystallography_data[, i]), i]
d3 <- function(x) { # construct the mixture using the estimated parameters
mix.object$lambda[1]*dgamma(x, shape=mix.object$gamma.pars[1,1], 1/mix.object$gamma.pars[2,1]) +
mix.object$lambda[2]*dgamma(x, shape=mix.object$gamma.pars[1,2], 1/mix.object$gamma.pars[2,2]) +
mix.object$lambda[3]*dgamma(x, shape=mix.object$gamma.pars[1,3], 1/mix.object$gamma.pars[2,3])
}
x <- seq(min(data_no_NA), max(data_no_NA), 0.001)
hist(data_no_NA, col="pink", freq=F, breaks=10, main = main, xlab="Intensities")
lines(x, d3(x), lwd=3, col="black", xlim=c(4,23), ylim=c(0, 0.25))
mixColors <- colorRampPalette(c("blue", "red"))(k)
for (i in 1:k) {
d = function(x) { # construct each of the Gamma components using the estimated parameters
mix.object$lambda[i]*dgamma(x, shape=mix.object$gamma.pars[1, i], 1/mix.object$gamma.pars[2,i])
}
lines(x, d(x), lwd=3, col=mixColors[i])
}
}
# Custom design of Weibull-Mixture Model plot
weibullMM.plot <- function(mix.object, k = 2, main = "") { # mix.object <- fit_W[[i]]
data_no_NA <- crystallography_data[complete.cases(crystallography_data[, i]), i]
d3 <- function(x) { # construct the mixture using the estimated parameters
mix.object$lambda[1]*dweibull(x, shape=mix.object$shape[1], scale=mix.object$scale[1]) +
mix.object$lambda[2]*dweibull(x, shape=mix.object$shape[2], scale=mix.object$scale[2]) +
mix.object$lambda[3]*dweibull(x, shape=mix.object$shape[3], scale=mix.object$scale[3])
}
x <- seq(min(data_no_NA), max(data_no_NA), 0.001)
hist(data_no_NA, col="pink", freq=F, breaks=15, main = main, xlab="Intensities")
lines(x, d3(x), lwd=3, col="black", xlim=c(4,23), ylim=c(0, 0.25))
mixColors <- colorRampPalette(c("blue", "red"))(k)
for (i in 1:k) {
d = function(x) { # construct each of the Weibull components using the estimated parameters
mix.object$lambda[i]*dweibull(x, shape=mix.object$shape[i], scale=mix.object$scale[i])
}
lines(x, d(x), lwd=3, col=mixColors[i])
}
}
# Custom design of Normal-Mixture Model plot
normalMM.plot <- function(mix.object, k = 2, main = "") { # mix.object <- fit_LN[[i]]
data_no_NA <- crystallography_data[complete.cases(crystallography_data[, i]), i]
d3 <- function(x) { # construct the mixture using the estimated parameters
mix.object$lambda[1]*dnorm(x, mean=mix.object$mu[1], sd=mix.object$sigma[1]) +
mix.object$lambda[2]*dnorm(x, mean=mix.object$mu[2], sd=mix.object$sigma[2]) +
mix.object$lambda[3]*dnorm(x, mean=mix.object$mu[3], sd=mix.object$sigma[3])
}
x <- seq(min(data_no_NA), max(data_no_NA), 0.001)
hist(data_no_NA, col="pink", freq=F, breaks=20, main = main, xlab="Intensities", xlim = c(4,180), ylim = c(0.0, 0.02))
lines(x, d3(x), lwd=3, col="black")
mixColors <- colorRampPalette(c("blue", "red"))(k)
for (i in 1:k) {
d = function(x) { # construct each of the Normal components using the estimated parameters
mix.object$lambda[i]*dnorm(x, mean=mix.object$mu[i], sd=mix.object$sigma[i])
}
lines(x, d(x), lwd=3, col=mixColors[i])
}
}Next, we will display the three alternative mixture distribution models overlaid on the sample histograms of each of the nine samples.
# Plot Mixture Models and Report model parameter estimates
# for(i in 1:col_num) { # uncomment this to plot all 9 samples
for(i in 1:2) { # this only plots the first 2 samples to save space
weibullMM.plot(fit_W[[i]], df_sampleMixtureParam[1,i],
paste0("Mixture of ", df_sampleMixtureParam[1, sampleColNames[i]],
" Weibull Models of ", sampleColNames[i]))
#plot(fit_W[[i]], density=TRUE, whichplots = 2,
# main2=paste0("Mixture of ", df_sampleMixtureParam[1, sampleColNames[i]],
# " Weibull Models of ", sampleColNames[i]), xlab2="Intensities")
gammaMM.plot(fit_G[[i]], df_sampleMixtureParam[1,i],
paste0("Mixture of ", df_sampleMixtureParam[1, sampleColNames[i]],
" Gamma Models of ", sampleColNames[i]))
normalMM.plot(fit_LN[[i]], df_sampleMixtureParam[1,i],
paste0("Mixture of ", df_sampleMixtureParam[1, sampleColNames[i]],
" Normal Models of ", sampleColNames[i]))
}





1.6.1.9 Reporting model parameter estimates
For each of the 9 samples in this dataset) and each of the 3 types of mixture distribution models (Weibull, Gamma, and Normal) we will summarize:
- lambda: The weights (impacts) of each of the 3 mixture components to the overall mixture model,
- parameters: of each mixture distribution component, mean and sd,
- loglik: the overall mixture distribution log-likelihood value.
# Generate the summary DF
getSummaryTable <- function (crystalSampleIndex) {
mat <- matrix(0, nrow = 3, ncol = 10)
# Weibull estimates for all 3 model components
# For Weibull Dist mean and SD see: https://en.wikipedia.org/wiki/Weibull_distribution
mat[1,1] <- round(fit_W[[crystalSampleIndex]]$lambda[1],3) # lambda
mat[1,2] <- round(fit_W[[crystalSampleIndex]]$scale[1] *
gamma(1+1/fit_W[[crystalSampleIndex]]$shape[1]),3) # mean
mat[1,3] <- round(fit_W[[crystalSampleIndex]]$scale[1] *
sqrt(gamma(1+2/fit_W[[crystalSampleIndex]]$shape[1])-
(gamma(1+1/fit_W[[crystalSampleIndex]]$shape[1]))^2),3) # sd
mat[1,4] <- round(fit_W[[crystalSampleIndex]]$lambda[2],3) # lambda
mat[1,5] <- round(fit_W[[crystalSampleIndex]]$scale[2] *
gamma(1+1/fit_W[[crystalSampleIndex]]$shape[2]),3) # mean
mat[1,6] <- round(fit_W[[crystalSampleIndex]]$scale[2] *
sqrt(gamma(1+2/fit_W[[crystalSampleIndex]]$shape[2])-
(gamma(1+1/fit_W[[crystalSampleIndex]]$shape[2]))^2),3) # sd
mat[1,7] <- round(fit_W[[crystalSampleIndex]]$lambda[3],3) # lambda
mat[1,8] <- round(fit_W[[crystalSampleIndex]]$scale[3] *
gamma(1+1/fit_W[[crystalSampleIndex]]$shape[3]),3) # mean
mat[1,9] <- round(fit_W[[crystalSampleIndex]]$scale[3] *
sqrt(gamma(1+2/fit_W[[crystalSampleIndex]]$shape[3])-
(gamma(1+1/fit_W[[crystalSampleIndex]]$shape[3]))^2),3) # sd
mat[1,10] <- round(fit_W[[crystalSampleIndex]]$loglik,3) # Log-lik
# Gamma estimates for all 3 model components
# For Gamma dist mean & SD see: https://en.wikipedia.org/wiki/Gamma_distribution
mat[2,1] <- round(fit_G[[crystalSampleIndex]]$lambda[1],3) # lambda
mat[2,2] <- round(fit_G[[crystalSampleIndex]]$gamma.pars[1,1]*
fit_G[[crystalSampleIndex]]$gamma.pars[2,1],3) # mean
mat[2,3] <- round(sqrt(fit_G[[crystalSampleIndex]]$gamma.pars[1,1])*
fit_G[[crystalSampleIndex]]$gamma.pars[2,1],3) # SD
mat[2,4] <- round(fit_G[[crystalSampleIndex]]$lambda[2],3) # lambda
mat[2,5] <- round(fit_G[[crystalSampleIndex]]$gamma.pars[1,2]*
fit_G[[crystalSampleIndex]]$gamma.pars[2,2],3) # mean
mat[2,6] <- round(sqrt(fit_G[[crystalSampleIndex]]$gamma.pars[1,2])*
fit_G[[crystalSampleIndex]]$gamma.pars[2,2],3) # sd
mat[2,7] <- round(fit_G[[crystalSampleIndex]]$lambda[3],3) # lambda
mat[2,8] <- round(fit_G[[crystalSampleIndex]]$gamma.pars[1,3]*
fit_G[[crystalSampleIndex]]$gamma.pars[2,3],3) # mean
mat[2,9] <- round(sqrt(fit_G[[crystalSampleIndex]]$gamma.pars[1,3])*
fit_G[[crystalSampleIndex]]$gamma.pars[2,3],3) # sd
mat[2,10] <- round(fit_G[[crystalSampleIndex]]$loglik,3) # Log-lik
# Normal estimates for all 3 model components
mat[3,1] <- round(fit_LN[[crystalSampleIndex]]$lambda[1],3) # lambda
mat[3,2] <- round(fit_LN[[crystalSampleIndex]]$mu[1],3) # shape
mat[3,3] <- round(fit_LN[[crystalSampleIndex]]$sigma[1],3) # scale
mat[3,4] <- round(fit_LN[[crystalSampleIndex]]$lambda[2],3) # lambda
mat[3,5] <- round(fit_LN[[crystalSampleIndex]]$mu[2],3) # shape
mat[3,6] <- round(fit_LN[[crystalSampleIndex]]$sigma[2],3) # scale
mat[3,7] <- round(fit_LN[[crystalSampleIndex]]$lambda[3],3) # lambda
mat[3,8] <- round(fit_LN[[crystalSampleIndex]]$mu[3],3) # shape
mat[3,9] <- round(fit_LN[[crystalSampleIndex]]$sigma[3],3) # scale
mat[3,10] <- round(fit_LN[[crystalSampleIndex]]$loglik,3) # Log-lik
return(as.data.frame(mat))
}
# render the summary DT tables
library("DT")Below we summarize the mixture-distribution models just for the first two crystallographic features.
1.6.1.9.1 AC1338 Report (Case 1)
1.6.1.9.2 AC1432 Report (Case 2)
1.6.2 2D Kernel Density and 3D Surface Plots
Density estimation is the process of using observed data to compute an estimate of the underlying process’ probability density function. There are several approaches to obtain density estimation, but the most basic technique is to use a rescaled histogram.
Plotting 2D Kernel Density and 3D Surface plots is very important and useful in multivariate exploratory data analytics.
We will use the plot_ly() function in the
plotly package, which works with data frame objects.
To create a surface plot, we use two vectors: x and y with length m and n respectively. We also need a matrix: z of size \(m\times n\). This z matrix is created from matrix multiplication between x and y.
To plot the 2D Kernel Density estimation plot we will use the
eruptions data from the “Old Faithful” geyser in Yellowstone National
Park, Wyoming stored under geyser. Also,
kde2d() function is needed for 2D kernel density
estimation.
## [1] 0.8333333 0.9275510 1.0217687 1.1159864 1.2102041## [1] 43.00000 44.32653 45.65306 46.97959 48.30612## [,1] [,2] [,3] [,4] [,5]
## [1,] 9.068691e-13 4.238943e-12 1.839285e-11 7.415672e-11 2.781459e-10
## [2,] 1.814923e-12 8.473636e-12 3.671290e-11 1.477410e-10 5.528260e-10
## [3,] 3.428664e-12 1.599235e-11 6.920273e-11 2.780463e-10 1.038314e-09
## [4,] 6.114498e-12 2.849475e-11 1.231748e-10 4.942437e-10 1.842547e-09
## [5,] 1.029643e-11 4.793481e-11 2.070127e-10 8.297218e-10 3.088867e-09Here z=t(x)%*%y. Then we apply plot_ly to
the list kd using the with() function.

Note we used the option "surface".
For 3D surfaces, we have a built-in dataset in R called
volcano. It records the volcano height at location x, y
(longitude, latitude). Because z is always made from x
and y, we can simply specify z to get the complete
surface plot.
## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
## [1,] 100 100 101 101 101 101 101 100 100 100
## [2,] 101 101 102 102 102 102 102 101 101 101
## [3,] 102 102 103 103 103 103 103 102 102 102
## [4,] 103 103 104 104 104 104 104 103 103 103
## [5,] 104 104 105 105 105 105 105 104 104 103
## [6,] 105 105 105 106 106 106 106 105 105 104
## [7,] 105 106 106 107 107 107 107 106 106 105
## [8,] 106 107 107 108 108 108 108 107 107 106
## [9,] 107 108 108 109 109 109 109 108 108 107
## [10,] 108 109 109 110 110 110 110 109 109 108
1.6.3 Multiple 2D image surface plots
#install.packages("jpeg") ## if necessary
library(jpeg)
# Get an image file downloaded (default: MRI_ImageHematoma.jpg)
img_url <- "https://umich.instructure.com/files/1627149/download?download_frd=1"
img_file <- tempfile(); download.file(img_url, img_file, mode="wb")
img <- readJPEG(img_file)
file.info(img_file)## size isdir
## C:\\Users\\IvoD\\AppData\\Local\\Temp\\RtmpSWuVNf\\file943471dc6999 8019 FALSE
## mode
## C:\\Users\\IvoD\\AppData\\Local\\Temp\\RtmpSWuVNf\\file943471dc6999 666
## mtime
## C:\\Users\\IvoD\\AppData\\Local\\Temp\\RtmpSWuVNf\\file943471dc6999 2026-04-29 12:19:18
## ctime
## C:\\Users\\IvoD\\AppData\\Local\\Temp\\RtmpSWuVNf\\file943471dc6999 2026-04-29 12:19:17
## atime
## C:\\Users\\IvoD\\AppData\\Local\\Temp\\RtmpSWuVNf\\file943471dc6999 2026-04-29 12:19:18
## exe
## C:\\Users\\IvoD\\AppData\\Local\\Temp\\RtmpSWuVNf\\file943471dc6999 no## [1] TRUEimg <- img[, , 1] # extract the first channel (from RGB intensity spectrum) as a univariate 2D array
# install.packages("spatstat")
# package spatstat has a function blur() that applies a Gaussian blur
library(spatstat)
img_s <- as.matrix(blur(as.im(img), sigma=10)) # the smoothed version of the image
z2 <- img_s + 1 # abs(rnorm(1, 1, 1)) # Upper confidence surface
z3 <- img_s - 1 # abs(rnorm(1, 1, 1)) # Lower confidence limit
# Plot the image surfaces
p <- plot_ly(z=img, type="surface", showscale=FALSE) %>%
add_trace(z=z2, type="surface", showscale=FALSE, opacity=0.98) %>%
add_trace(z=z3, type="surface", showscale=FALSE, opacity=0.98)
p # Plot the mean-surface along with lower and upper confidence services.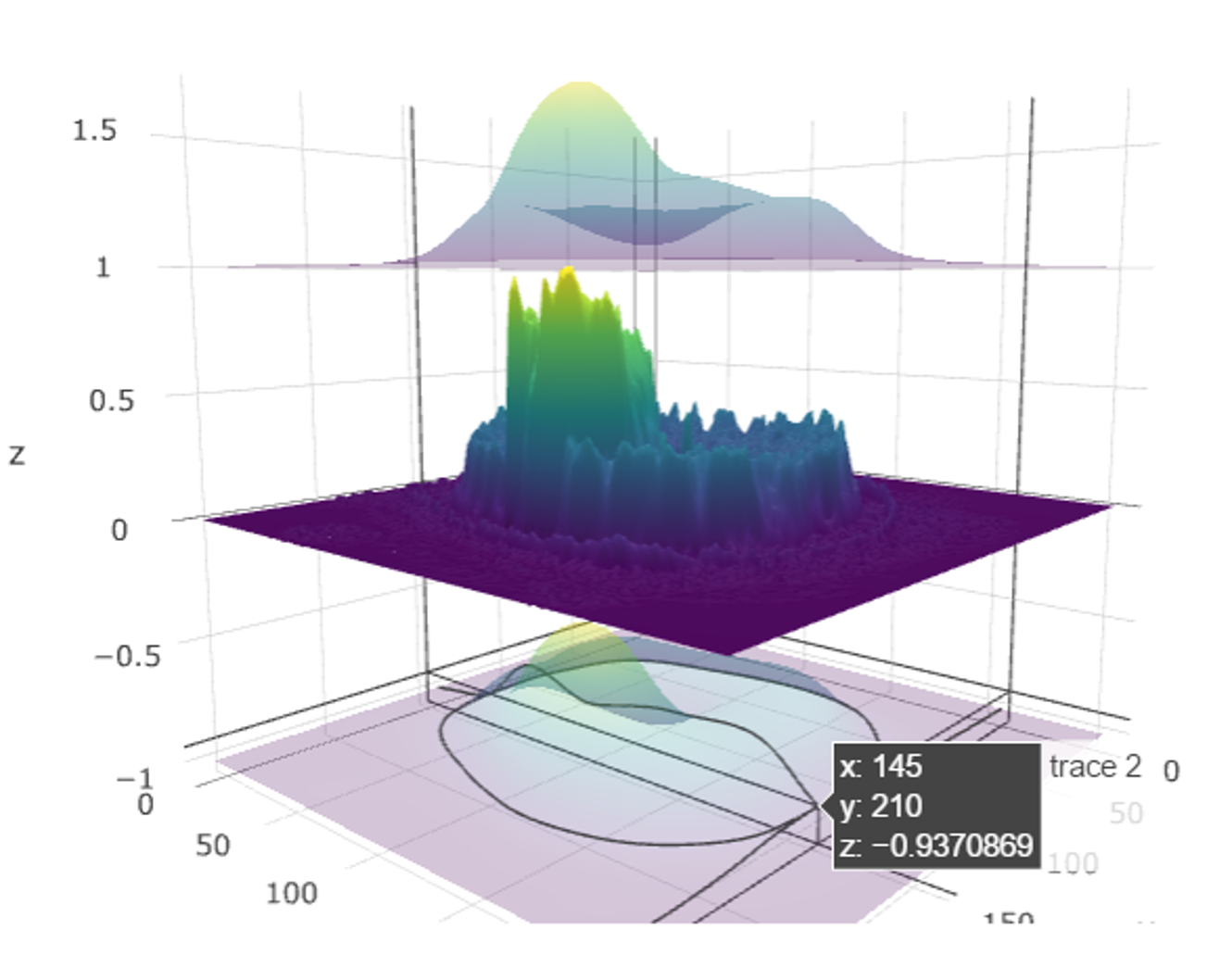
1.6.4 3D and 4D Visualizations
Many datasets have intrinsic multi-dimensional characteristics. For instance, the human body is a 3D solid of matter (3 spatial dimensions can be used to describe the position of every component, e.g., sMRI volume) that changes over time (the fourth dimension, e.g., fMRI hypervolumes).
The SOCR BrainViewer shows how to use a web-browser to visualize 2D cross-sections of 3D volumes, display volume-rendering, and show 1D (e.g., 1-manifold curves embedded in 3D) and 2D (e.g., surfaces, shapes) models jointly into the same 3D scene.
We will now illustrate an example of 3D/4D visualization in
R using the packages brainR
and rgl. This
code is included as it runs well in interactive R sessions.
However, it is suppressed during HTML knitting
(eval=FALSE), as rgl causes some browser-OS
combinations to fail while loading the resulting HTML file.
# install.packages("brainR") ## if necessary
library(brainR)
# Test data: https://socr.umich.edu/HTML5/BrainViewer/data/TestBrain.nii.gz
brainURL <- "https://socr.umich.edu/HTML5/BrainViewer/data/TestBrain.nii.gz"
brainFile <- file.path(tempdir(), "TestBrain.nii.gz")
download.file(brainURL, dest=brainFile, quiet=TRUE)
brainVolume <- readNIfTI(brainFile, reorient=FALSE)
brainVolDims <- dim(brainVolume); brainVolDims
# try different levels at which to construct contour surfaces (10 fast)
# lower values yield smoother surfaces # see ?contour3d
contour3d(brainVolume, level = 20, alpha = 0.1, draw = TRUE)
# multiple levels may be used to show multiple shells
# "activations" or surfaces like hyper-intense white matter
# This will take 1-2 minutes to rend!
contour3d(brainVolume, level = c(10, 120), alpha = c(0.3, 0.5),
add = TRUE, color=c("yellow", "red"))
# create text for orientation of right/left
text3d(x=brainVolDims[1]/2, y=brainVolDims[2]/2, z = brainVolDims[3]*0.98, text="Top")
text3d(x=brainVolDims[1]*0.98, y=brainVolDims[2]/2, z = brainVolDims[3]/2, text="Right")
### render this on a webpage and view it!
#browseURL(paste("file://",
# writeWebGL_split(dir= file.path(tempdir(),"webGL"),
# template = system.file("my_template.html", package="brainR"),
# width=500), sep=""))Below we provide some additional 3D/4D PET, sMRI, and fMRI volumes in *.nii.gz format:
- sMRI (3D real-valued structural MRI volume)
- fMRI (4D real-valued functional MRI hyper-volume)
- PET (3D perfusion Positron Emission Tomography volume).
For 4D fMRI time-series, we can load the hypervolumes similarly and then display some lower dimensional projections.
# See examples here: https://cran.r-project.org/web/packages/oro.nifti/vignettes/nifti.pdf
# and here: http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0089470
library(oro.nifti)
fMRIURL <- "https://socr.umich.edu/HTML5/BrainViewer/data/fMRI_FilteredData_4D.nii.gz"
fMRIFile <- file.path(tempdir(), "fMRI_FilteredData_4D.nii.gz")
download.file(fMRIURL, dest=fMRIFile, quiet=TRUE)
(fMRIVolume <- readNIfTI(fMRIFile, reorient=FALSE))## NIfTI-1 format
## Type : nifti
## Data Type : 4 (INT16)
## Bits per Pixel : 16
## Slice Code : 0 (Unknown)
## Intent Code : 0 (None)
## Qform Code : 1 (Scanner_Anat)
## Sform Code : 0 (Unknown)
## Dimension : 64 x 64 x 21 x 180
## Pixel Dimension : 4 x 4 x 6 x 3
## Voxel Units : mm
## Time Units : sec# dimensions: 64 x 64 x 21 x 180 ; 4mm x 4mm x 6mm x 3 sec
fMRIVolDims <- dim(fMRIVolume); fMRIVolDims## [1] 64 64 21 180## [1] 180# Plot the 4D array of imaging data in a 5x5 grid of images
# The first three dimensions are spatial locations of the voxel (volume element) and the fourth dimension is time for this functional MRI (fMRI) acquisition.
image(fMRIVolume, zlim=range(fMRIVolume)*0.95)
h <- hist(fMRIVolume, plot = F)
plot_ly(x = h$mids, y = h$density, type = "bar") %>%
layout(bargap=0.1, title="fMRI Histogram")# Plot an orthographic display of the fMRI data using the axial plane containing the left-and-right thalamus to approximately center the crosshair vertically
orthographic(fMRIVolume, xyz=c(34,29,10), zlim=range(fMRIVolume)*0.9)
stat_fmri_test <- ifelse(fMRIVolume > 15000, fMRIVolume, NA)
h <- hist(stat_fmri_test, plot = F)
plot_ly(x = h$mids, y = h$density, type = "bar") %>%
layout(bargap=0.1, title="fMRI Histogram (high intensities)")## [1] 64 64 21 180
# overlay(fMRIVolume, stat_fmri_test[,,,5], zlim.x=range(fMRIVolume)*0.95)
# To examine the time course of a specific 3D voxel (say the one at x=30, y=30, z=10):
# plot(fMRIVolume[30, 30, 10,], type='l', main="Time Series of 3D Voxel \n (x=30, y=30, z=10)", col="blue")
x1 <- c(1:180)
y1 <- loess(fMRIVolume[30, 30, 10,]~ x1, family = "gaussian")
# lines(x1, smooth(fMRIVolume[30, 30, 10,]), col = "red", lwd = 2)
# lines(ksmooth(x1, fMRIVolume[30, 30, 10,], kernel = "normal", bandwidth = 5), col = "green", lwd = 3)
# legend("bottomright", legend=c("(raw) fMRI", "smooth(fMRI)", "ksmooth(fMRI"),
# col=c("blue", "red", "green"), lty=1, cex=0.8,
# y.intersp=0.8)
plot_ly(x = x1, y = fMRIVolume[30, 30, 10,],
name="Raw fMRI", type = 'scatter', mode = 'lines') %>%
add_trace(y = smooth(fMRIVolume[30, 30, 10,]), name = 'loess fMRI') %>%
add_trace(y = ksmooth(x1, fMRIVolume[30, 30, 10,], kernel="normal", bandwidth = 5)$y, name='kSmooth fMRI') %>%
layout(title="Time Series of 3D Voxel (x=30, y=30, z=10)", legend = list(orientation = 'h'))Chapter 12 provides more details about longitudinal and time-series data analysis.
Finally, DSPA Appendix 3 includes details about classification, representation, modeling, and visualization of parametric and implicit, open and closed manifolds.
2 Appendix
2.1 Importing Data from SQL Databases
Review DSPA
Appendix 5 for more details on DB access. We can also import SQL
databases into R. First, we need to install and load the
RODBC (R Open Database Connectivity) package.
Then, we could open a connection to the SQL server database with Data Source Name (DSN), via Microsoft Access. More details are provided here and here.
2.2 Additional
R scripts
The code below was used to generate some of the graphs shown in this chapter.
# Right Skewed
N <- 10000
x <- rnbinom(N, 10, .5)
hist(x,
xlim=c(min(x), max(x)), probability=T, nclass=max(x)-min(x)+1,
col='lightblue', xlab=' ', ylab=' ', axes=F,
main='Right Skewed')
lines(density(x, bw=1), col='red', lwd=3)
#No Skew
N <- 10000
x <- rnorm(N, 0, 1)
hist(x, probability=T,
col='lightblue', xlab=' ', ylab=' ', axes=F,
main='No Skew')
lines(density(x, bw=0.4), col='red', lwd=3)
#Uniform density
x<-runif(1000, 1, 50)
hist(x, col='lightblue', main="Uniform Distribution", probability = T, xlab="", ylab="Density", axes=F)
abline(h=0.02, col='red', lwd=3)
#68-95-99.7 rule
x <- rnorm(N, 0, 1)
hist(x, probability=T,
col='lightblue', xlab=' ', ylab=' ', axes = F,
main='68-95-99.7 Rule')
lines(density(x, bw=0.4), col='red', lwd=3)
axis(1, at=c(-3, -2, -1, 0, 1, 2, 3), labels = expression(mu-3*sigma, mu-2*sigma, mu-sigma, mu, mu+sigma, mu+2*sigma, mu+3*sigma))
abline(v=-1, lwd=3, lty=2)
abline(v=1, lwd=3, lty=2)
abline(v=-2, lwd=3, lty=2)
abline(v=2, lwd=3, lty=2)
abline(v=-3, lwd=3, lty=2)
abline(v=3, lwd=3, lty=2)
text(0, 0.2, "68%")
segments(-1, 0.2, -0.3, 0.2, col = 'red', lwd=2)
segments(1, 0.2, 0.3, 0.2, col = 'red', lwd=2)
text(0, 0.15, "95%")
segments(-2, 0.15, -0.3, 0.15, col = 'red', lwd=2)
segments(2, 0.15, 0.3, 0.15, col = 'red', lwd=2)
text(0, 0.1, "99.7%")
segments(-3, 0.1, -0.3, 0.1, col = 'red', lwd=2)
segments(3, 0.1, 0.3, 0.1, col = 'red', lwd=2)2.3 Case-Study 11 - Traumatic Brain Injury (TBI)
The data is available in the Canvas case-studies folder.
# load data CaseStudy11_TBI.xlsx
tmp = tempfile(fileext = ".xlsx")
download.file(url = "https://umich.instructure.com/files/416270/download?download_frd=1", destfile = tmp, mode="wb")
df_TBI <- openxlsx::read.xlsx(xlsxFile = tmp, sheet = "Sheet1", skipEmptyRows = TRUE)
dim(df_TBI)## [1] 46 19Preprocess the data and plot the clustering dendrogram.
# install.packages("dendextend")
library(dendextend)
# Clean the data first (missing values, characters, etc.)
na_strings <- c("NA", ".")
df_TBI_clean <- df_TBI %>% naniar::replace_with_na_all(condition = ~.x %in% na_strings)
df_TBI_clean <- as.data.frame(df_TBI_clean[, -c(3:4)])
df_TBI_clean <- df_TBI_clean %>% tidyr::drop_na ()
dim(df_TBI_clean) # [1] 23 17## [1] 23 17rownames(df_TBI_clean) <- as.character(df_TBI_clean[ ,1])
df_TBI_clean <- df_TBI_clean[, -1]
df_TBI_clean <- as.data.frame(sapply(df_TBI_clean, as.numeric))
df_TBI_clean <- df_TBI_clean[, c("age", "2013.gose", "skull.fx", "temp.injury", "surgery", "acute.sz")]
df_TBI_clean <- as.data.frame(scale(df_TBI_clean))
hc <- hclust(dist(df_TBI_clean), "ave")
dend <- as.dendrogram(hc)
plot_dendro(dend, height = 600) %>%
layout(xaxis = list(range = c(-1, 5))) %>%
hide_legend() %>%
highlight(persistent = TRUE, dynamic = TRUE)# cutree(hc, k = 2)
# alternatively specify the height, which is, the value of the criterion associated with the
# clustering method for the particular agglomeration -- cutree(hc, h= 10)
table(cutree(hc, h= 3)) # cluster distribution##
## 1 2 3 4 5 6
## 6 10 1 3 1 2To identify the number of cases for varying number of clusters
# To identify the number of cases for varying number of clusters we can combine calls to cutree and table
# in a call to sapply -- to see the sizes of the clusters for $2\ge k \ge 10$ cluster-solutions:
# numbClusters=4;
myClusters = sapply(2:5, function(numbClusters)table(cutree(hc, numbClusters)))
names(myClusters) <- paste("Number of Clusters=", 2:5, sep = "")
myClusters## $`Number of Clusters=2`
##
## 1 2
## 19 4
##
## $`Number of Clusters=3`
##
## 1 2 3
## 6 13 4
##
## $`Number of Clusters=4`
##
## 1 2 3 4
## 6 11 4 2
##
## $`Number of Clusters=5`
##
## 1 2 3 4 5
## 6 11 3 1 2Inspect which SubjectIDs are in which clusters:
##
## 1 2
## 19 4groups.k.2 <- cutree(hc, k = 2)
sapply(unique(groups.k.2), function(g) rownames(df_TBI_clean)[groups.k.2 == g])## [[1]]
## [1] "1" "2" "3" "4" "5" "6" "7" "8" "9" "11" "12" "14" "15" "16" "17"
## [16] "18" "19" "20" "21"
##
## [[2]]
## [1] "10" "13" "22" "23"Let’s see which Age and which Surgery cohorts fall within each of the derived cluster labels. Remember that all variables are scaled, so they represent standardized variable values!
groups.k.3 <- cutree(hc, k = 3)
sapply(unique(groups.k.3), function(g) df_TBI_clean$age[groups.k.3 == g])## [[1]]
## [1] -0.8625007 0.3227597 -0.4258258 -1.2367934 -1.1744113 0.6346703
##
## [[2]]
## [1] 1.19610942 1.00896305 -1.36155766 -0.80011855 -0.48820793 0.01084907
## [7] 0.13561331 -0.98726492 -0.23867943 2.44375190 1.50802004 0.19799544
## [13] 1.38325579
##
## [[3]]
## [1] -0.1762973 -1.0496470 0.1979954 -0.2386794## [[1]]
## [1] -1.219804 0.784160 -1.219804 -1.219804 -1.219804 0.784160
##
## [[2]]
## [1] 0.784160 0.784160 0.784160 -1.219804 0.784160 0.784160 0.784160
## [8] 0.784160 0.784160 -1.219804 0.784160 0.784160 -1.219804
##
## [[3]]
## [1] -1.219804 0.784160 -1.219804 0.784160# Note that there may be dependencies between some variables
fit <- lm(`2013.gose` ~ age, data = df_TBI_clean)
plot_ly(df_TBI_clean, x = ~age, y = ~`2013.gose`, type = 'scatter', mode = "markers", name="Data") %>%
add_lines(x = ~age, y = fit$fitted.values, mode = "lines", name="Linear Model") %>%
layout(title=paste0("Correlation(2013.gose,age) = ", round(cor(df_TBI_clean$`2013.gose`, df_TBI_clean$age),3)))##
## groups.k.3 -1.21980437173918 0.7841599532609
## 1 4 2
## 2 3 10
## 3 2 2To characterize the clusters, we can look at cluster summary statistics, like the median, of the variables that were used to perform the cluster analysis. These can be broken down by the groups identified by the cluster analysis. The aggregate function will compute stats (e.g., median) on many variables simultaneously. To look at the median values for the variables we’ve used in the cluster analysis, broken up by the cluster groups:
## Group.1 age 2013.gose skull.fx temp.injury surgery acute.sz
## 1 1 -0.6441632 0.7779885 -0.2178222 -1.646252 -1.2198044 -0.448746
## 2 2 0.1356133 -0.1637871 0.7841600 0.581030 0.7841600 -0.448746
## 3 3 -0.2074884 -0.1637871 0.7841600 0.581030 -0.2178222 2.1315442.4 Some additional
ggplot examples
2.4.1 Housing Price Data
This example uses the SOCR Home Price Index data of 19 major city in US from 1991-2009.
library(rvest)
# draw data
wiki_url <- read_html("https://wiki.socr.umich.edu/index.php/SOCR_Data_Dinov_091609_SnP_HomePriceIndex")
hm_price_index<- html_table(html_nodes(wiki_url, "table")[[1]])
head(hm_price_index)## # A tibble: 6 × 23
## Index Year Month `AZ-Phoenix` `CA-LosAngeles` `CA-SanDiego` `CA-SanFrancisco`
## <int> <int> <chr> <dbl> <dbl> <dbl> <dbl>
## 1 1 1991 Janu… 65.3 95.3 83.1 71.2
## 2 2 1991 Febr… 65.3 94.1 81.9 70.3
## 3 3 1991 March 64.6 92.8 80.9 69.6
## 4 4 1991 April 64.4 92.8 80.7 69.5
## 5 5 1991 May 64.4 93.4 81.4 70.1
## 6 6 1991 June 64.9 94.2 82.2 70.8
## # ℹ 16 more variables: `CO-Denver` <dbl>, `DC-Washington` <dbl>,
## # `FL-Miami` <dbl>, `FL-Tampa` <dbl>, `GA-Atlanta` <dbl>, `IL-Chicago` <dbl>,
## # `MA-Boston` <dbl>, `MI-Detroit` <dbl>, `MN-Minneapolis` <dbl>,
## # `NC-Charlotte` <dbl>, `NV-LasVegas` <dbl>, `NY-NewYork` <dbl>,
## # `OH-Cleveland` <dbl>, `OR-Portland` <dbl>, `WA-Seattle` <dbl>,
## # `Composite-10` <dbl>period <- lubridate::parse_date_time(paste(hm_price_index$Year, hm_price_index$Month), "ym")
hm_price_index <- hm_price_index[, c(-1,-2, -3)]
hm_price_index$Date <- period
library(reshape2)
hm_index_melted = melt(hm_price_index, id.vars='Date') #a common trick for plot, wide -> long format
# ggplot(data=hm_index_melted, aes(x=Date, y=value, color=variable)) +
# geom_line(size=1.5) + ggtitle("HomePriceIndex:1991-2009")
plot_ly(hm_index_melted, x=~Date, y=~value, color=~variable,
type="scatter", mode="lines+markers") %>%
layout(title="US Housing Price Index (1991-2009)", yaxis=list(title="HPI"), legend=list(orientation = 'h'))2.4.2 Modeling the home price index data
#Linear regression and predict
hm_price_index$pred = predict(lm(`CA-SanFrancisco` ~ `CA-LosAngeles`, data=hm_price_index))
# ggplot(data=hm_price_index, aes(x = `CA-LosAngeles`)) +
# geom_point(aes(y = `CA-SanFrancisco`)) +
# geom_line(aes(y = pred), color='Magenta', size=2) + ggtitle("PredictHomeIndex SF - LA")
plot_ly(hm_price_index, x=~`CA-LosAngeles`, y=~`CA-SanFrancisco`, color=~`Composite-10`,
type="scatter", mode="lines+markers", name="HPI Data") %>%
add_lines(x = ~`CA-LosAngeles`, y = hm_price_index$pred, mode = "lines", name="Linear Model") %>%
layout(title="LA (SoCal) vs. FS (NoCal)", yaxis=list(title="Los Angeles"),
yaxis=list(title="San Francisco"), legend=list(orientation = 'h'))## Warning: line.color doesn't (yet) support data arrays
## Warning: line.color doesn't (yet) support data arrays
## Warning: line.color doesn't (yet) support data arrays
## Warning: line.color doesn't (yet) support data arraysLet’s examine some popular ggplot graphs.
## # A tibble: 6 × 6
## `IL-Chicago` `MA-Boston` `MI-Detroit` `MN-Minneapolis` `NC-Charlotte`
## <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 70.0 65.0 58.2 64.2 73.3
## 2 70.5 64.2 57.8 64.2 73.3
## 3 70.6 63.6 57.6 64.2 72.8
## 4 71.1 63.4 57.8 64.3 72.9
## 5 71.4 63.8 58.4 64.8 73.3
## 6 71.7 64.2 58.9 65.0 73.5
## # ℹ 1 more variable: `NV-LasVegas` <dbl>colnames(pairs) <- c("Atlanta", "Chicago", "Boston", "Detroit", "Minneapolis", "Charlotte")
ggpairs(pairs) # you can define the plot design by specifying "upper", "lower", "diag", etc. 
2.4.3 Map of the neighborhoods of Los Angeles (LA)
This example interrogates data of 110 LA neighborhoods, which includes measures of education, income and population demographics.
Here, we select the Longitude and Latitude as the axes, mark these 110 Neighborhoods according to their population, fill out those points according to the income of each area, and label each neighborhood.
library(rvest)
library(ggplot2)
#draw data
wiki_url <- read_html("https://wiki.socr.umich.edu/index.php/SOCR_Data_LA_Neighborhoods_Data")
html_nodes(wiki_url, "#content")## {xml_nodeset (1)}
## [1] <div id="content" class="mw-body" role="main">\n\t\t\t<a id="top"></a>\n\ ...LA_Nbhd_data <- html_table(html_nodes(wiki_url, "table")[[2]])
#display several lines of data
head(LA_Nbhd_data); ## # A tibble: 6 × 15
## LA_Nbhd Income Schools Diversity Age Homes Vets Asian Black Latino White
## <chr> <int> <int> <dbl> <int> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 Adams_Nor… 29606 691 0.6 26 0.26 0.05 0.05 0.25 0.62 0.06
## 2 Arleta 65649 719 0.4 29 0.29 0.07 0.11 0.02 0.72 0.13
## 3 Arlington… 31423 687 0.8 31 0.31 0.05 0.13 0.25 0.57 0.05
## 4 Atwater_V… 53872 762 0.9 34 0.34 0.06 0.2 0.01 0.51 0.22
## 5 Baldwin_H… 37948 656 0.4 36 0.36 0.1 0.05 0.71 0.17 0.03
## 6 Bel-Air 208861 924 0.2 46 0.46 0.13 0.08 0.01 0.05 0.83
## # ℹ 4 more variables: Population <int>, Area <dbl>, Longitude <dbl>,
## # Latitude <dbl>theme_set(theme_grey())
#treat ggplot as a variable
#When claim "data", we can access its column directly e.g., "x = Longitude"
plot1 = ggplot(data=LA_Nbhd_data, aes(x=LA_Nbhd_data$Longitude, y=LA_Nbhd_data$Latitude))
#you can easily add attribute, points, label(e.g., :text)
plot1 + geom_point(aes(size=Population, fill=LA_Nbhd_data$Income), pch=21, stroke=0.2, alpha=0.7, color=2)+
geom_text(aes(label=LA_Nbhd_data$LA_Nbhd), size=1.5, hjust=0.5, vjust=2, check_overlap = T)+
scale_size_area() + scale_fill_distiller(limits=c(range(LA_Nbhd_data$Income)), palette='RdBu', na.value='white', name='Income') +
scale_y_continuous(limits=c(min(LA_Nbhd_data$Latitude), max(LA_Nbhd_data$Latitude))) +
coord_fixed(ratio=1) + ggtitle('LA Neighborhoods Scatter Plot (Location, Population, Income)') 
Observe that some areas (e.g., Beverly Hills) have disproportionately higher incomes and notice that the resulting plot resembles this plot
 .
.
2.4.4 Latin letter frequency in different languages
This example uses ggplot to interrogate the SOCR
Latin letter frequency data.
library(rvest)
wiki_url <- read_html("https://wiki.socr.umich.edu/index.php/SOCR_LetterFrequencyData")
letter<- html_table(html_nodes(wiki_url, "table")[[1]])
summary(letter)## Letter English French German
## Length:27 Min. :0.00000 Min. :0.00000 Min. :0.00000
## Class :character 1st Qu.:0.01000 1st Qu.:0.01000 1st Qu.:0.01000
## Mode :character Median :0.02000 Median :0.03000 Median :0.03000
## Mean :0.03667 Mean :0.03704 Mean :0.03741
## 3rd Qu.:0.06000 3rd Qu.:0.06500 3rd Qu.:0.05500
## Max. :0.13000 Max. :0.15000 Max. :0.17000
## Spanish Portuguese Esperanto Italian
## Min. :0.00000 Min. :0.00000 Min. :0.00000 Min. :0.00000
## 1st Qu.:0.01000 1st Qu.:0.00500 1st Qu.:0.01000 1st Qu.:0.00500
## Median :0.03000 Median :0.03000 Median :0.03000 Median :0.03000
## Mean :0.03815 Mean :0.03778 Mean :0.03704 Mean :0.03815
## 3rd Qu.:0.06000 3rd Qu.:0.05000 3rd Qu.:0.06000 3rd Qu.:0.06000
## Max. :0.14000 Max. :0.15000 Max. :0.12000 Max. :0.12000
## Turkish Swedish Polish Toki_Pona
## Min. :0.00000 Min. :0.00000 Min. :0.00000 Min. :0.00000
## 1st Qu.:0.01000 1st Qu.:0.01000 1st Qu.:0.01500 1st Qu.:0.00000
## Median :0.03000 Median :0.03000 Median :0.03000 Median :0.03000
## Mean :0.03667 Mean :0.03704 Mean :0.03704 Mean :0.03704
## 3rd Qu.:0.05500 3rd Qu.:0.05500 3rd Qu.:0.04500 3rd Qu.:0.05000
## Max. :0.12000 Max. :0.10000 Max. :0.20000 Max. :0.17000
## Dutch Avgerage
## Min. :0.00000 Min. :0.00000
## 1st Qu.:0.01000 1st Qu.:0.01000
## Median :0.02000 Median :0.03000
## Mean :0.03704 Mean :0.03741
## 3rd Qu.:0.06000 3rd Qu.:0.06000
## Max. :0.19000 Max. :0.12000## # A tibble: 6 × 14
## Letter English French German Spanish Portuguese Esperanto Italian Turkish
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 a 0.08 0.08 0.07 0.13 0.15 0.12 0.12 0.12
## 2 b 0.01 0.01 0.02 0.01 0.01 0.01 0.01 0.03
## 3 c 0.03 0.03 0.03 0.05 0.04 0.01 0.05 0.01
## 4 d 0.04 0.04 0.05 0.06 0.05 0.03 0.04 0.05
## 5 e 0.13 0.15 0.17 0.14 0.13 0.09 0.12 0.09
## 6 f 0.02 0.01 0.02 0.01 0.01 0.01 0.01 0
## # ℹ 5 more variables: Swedish <dbl>, Polish <dbl>, Toki_Pona <dbl>,
## # Dutch <dbl>, Avgerage <dbl>## [1] 13.08# require(reshape)
# library(scales)
# dtm = melt(letter[, -14], id.vars = c('Letter'))
# p = ggplot(dtm, aes(x = Letter, y = value, fill = variable)) +
# geom_bar(position = "fill", stat = "identity") +
# scale_y_continuous(labels = percent_format())+ggtitle('Pie Chart')
# #or exchange
# #p = ggplot(dtm, aes(x = variable, y = value, fill = Letter)) + geom_bar(position = "fill", stat = "identity") + scale_y_continuous(labels = percent_format())
# p
# #gg pie plot actually is stack plot + polar coordinate
# p + coord_polar()
reshape2::melt(letter, id.vars='Letter') %>%
plot_ly(x = ~Letter, y = ~value, type = 'bar',
name = ~variable, color = ~variable) %>%
layout(yaxis = list(title = 'Count'), barmode = 'stack')You can see some additional Latin Letters plots here.
Also review Visualization Chapter Part 1, which includes data handling, statistical measures of centrality and dispersion, understanding categorical and numeric data, uniform and normal distributions, missing data imputation, web page parsing, visualization of tabular HTML data, and cohort-rebalancing (for imbalanced groups).